
Journal of Chemistry and Applications
Download PDF
Case Study
*Address for Correspondence: Mohammad Asif, Department of Pharmacy, GRD (PG) Institute of Management & Technology, Dehradun, Uttarakhand, 248009, India, E-mail: aasif321@gmail.com
Citation: Asif M. A Mini Review on the Study of New Broad-Spectrum Antimicrobial Fluoroquinolone JNJ-Q2. J Chem Applications. 2014;1(1): 5.
Copyright © 2014 Asif M. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Chemistry & Applications | ISSN 2380-5021 | Volume: 1, Issue: 1
Submission: 03 April 2014 | Accepted: 29 May 2014 | Published: 04 June 2014
 Generations of Quinolones
Generations of Quinolones

 First-generation
First-generation
 The treatment of infections caused by drug-resistant Staphylococcus aureus (DRSA) particularly methicillin-resistant S. aureus (MRSA). Despite the emergence of resistant and MDR S. aureus, seven effective drugs in clinical use for which little resistance has been observed (vancomycin, quinupristin-dalfopristin, telavancin, ceftaroline, linezolid, tigecycline, and daptomycin). However, vancomycin is less effective for infections with MRSA isolates, and has a higher minimum inhibitory concentration (MIC) within the susceptible range. Linezolid is probably the drug of choice for the treatment of complicated MRSA skin and soft tissue infections (SSTIs). Whether drug of choice in pneumonia remains debatable. Daptomycin has shown to be non-inferior to either vancomycin or β-lactams in the treatment of staphylococcal SSTIs, bacteremia, and right-sided endocarditis. Tigecycline was also noninferior to comparator drugs in the treatment of SSTIs, but there is controversy about whether it is less effective than other therapeutic options in the treatment of more serious infections. Telavancin has been shown to be non-inferior to vancomycin in the treatment of SSTIs and pneumonia, but has greater nephrotoxicity. Ceftaroline is a broad-spectrum cephalosporin with activity against MRSA; it is non-inferior to vancomycin in the treatment of SSTIs. Clindamycin, trimethoprim-sulfamethoxazole, doxycycline, rifampin, moxifloxacin (MXF), and minocycline are oral anti-staphylococcal agents that may have utility in the treatment of SSTIs and osteomyelitis, but the clinical data for their efficacy is limited. There are also several drugs with broad-spectrum activity against Gram-positive organisms that have reached the phase II and III stages of clinical testing that will hopefully be approved for clinical use in the upcoming years: oritavancin, dalbavancin, omadacycline, tedizolid, delafloxacin, and JNJ-Q2. Thus, there are currently many effective drugs to treat resistant S. aureus infections and many promising agents in the pipeline. There are still frequent treatment failures and unfortunate clinical outcomes. The clinical challenges presented by MRSA, the clinical experience with current anti-MRSA antibiotics, and the gaps in knowledge on how to use these agents to most effectively combat MRSA infections [11]. JNJ-Q2 is a broad-spectrum FQL with activity against Gram-positive and Gram-negative pathogens. The in vitro activity of JNJ-Q2 was evaluated when tested against Neisseria gonorrhoeae isolates, including ciprofloxacin (CPFX)-resistant strains with documented mutations in the quinolone resistance determining region. MIC values were determined using CPFX, tetracycline, penicillin, ceftriaxone, and azithromycin as comparators. All isolates were inhibited by ≤ 0.25 μg/mL of JNJ-Q2 (range, 0.004-0.25 μg/mL; MIC which was 8-fold (MIC50) and 32-fold more potent (MIC90) compared to CPFX. Few strains were susceptible to penicillin (3.0%) and tetracycline (6.1%), and with the use of the Susceptibility Testing interpretive criteria, 13.6% were resistant to azithromycin. All strains were susceptible to ceftriaxone, the most potent agent (MIC90, 0.06 μg/mL) followed by JNJ-Q2 (MIC90, 0.25 μg/mL). JNJ-Q2 appears to be a potent FQL when tested against contemporary MDR N. gonorrhoeae [12]. JNJ-Q2, a fluorinated 4-quinolone, was very active against both methicillin-susceptible S. aureus (MSSA) and MRSA obtained from patients with acute bacterial skin and skin-structure infection (ABSSSI). Overall MXF and levofloxacin (LVFX) resistance rates were 31.5% and 46.9%, respectively. These favorable results support the continued clinical development of JNJ-Q2 to treat ABSSSI [13]. New antibiotic agents are desperately needed to treat the multidrugresistant (MDR) pathogens that continue to emerge at alarming rates. Many of the agents that have entered full clinical development since 1995 have been members of previously accepted classes of antibiotics. Among these are a new aminoglycoside (plazomicin), anti-MRSA cephalosporins (ceftobiprole, ceftaroline), a monocyclic β-lactam (BAL30072), the β-lactamase inhibitor combination of tazobactam with the anti-pseudomonal cephalosporin ceftolozane, β-lactam combinations with new non-β-lactam inhibitors (MK-7655 with imipenem, and avibactam with ceftazidime and ceftaroline), macrolides (cethromycin and solithromycin), oxazolidinones (tedizolid phosphate and radezolid), and quinolones (delafloxacin, nemonoxacin and JNJ-Q2). Resistance and safety issues have been circumvented by some of these new agents that have well-established mechanisms of action and defined pathways leading toward regulatory approval [14]. JNJ-Q2 is currently in clinical development for the treatment of community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin-structure infections. The JNJ-Q2 showed activity against common respiratory pathogens from patients with CABP. Streptococcus pneumonia (S. pneumoniae), Haemophilus influenzae and Moraxella catarrhalis were tested. JNJ-Q2 had activity against all three species, with 96.9% of strains inhibited at ≤ 0.015 mg/L. JNJ-Q2 [MIC for 50% and 90% of the organisms, respectively demonstrated a 16-fold greater potency compared with MFX and at least 128-fold greater activity compared with levofloxacin (LVFX) and CPFX against S. pneumoniae. The H. influenzae isolates were 21.9-23.3% resistant to ampicillin, but JNJ-Q2 was at least two-fold more active than MXF as well as being potent against M. catarrhalis. The JNJ-Q2 demonstrated increased potency compared with other marketed FQLs that have been used to treat CABP pathogens, thus favouring further clinical development [15]. JNJ-Q2, with anti-MRSA activity, was evaluated in a comprehensive set of non-clinical and clinical cardiovascular safety studies. The effect of JNJ-Q2 on different cardiovascular parameters was compared with that of MFX, sparfloxacin (SPFX) and ofloxacin. Through comparisons with these well-known FQLs, the importance of effects on compensatory ion channels to the cardiovascular safety of JNJ-Q2 was investigated. The trend for effects of JNJ-Q2 on Tp-Te, QT, QRS and PR intervals in the non-clinical models and the plateau in QTc with increasing plasma concentration in humans are consistent with offsetting sodium and calcium channel activities. Based on the non-clinical and clinical cardiovascular safety assessment, JNJ-Q2 has a safe cardiovascular profile for administration in humans with comparable or reduced potential to prolong QT intervals, compared with MFX. The importance of compensatory sodium and calcium channel activity in offsetting potassium channel activity for compounds with a FQL core [16]. JNJ-Q2 have a balanced potency against both DNA gyrase and topoisomerase IV targets. JNJ-Q2 is in the treatment of acute bacterial skin and skin-structure infections (ABSSSIs) and community-acquired bacterial pneumonia. MIC values were obtained for pathogens (44.4% were from patients diagnosed with ABSSSI). JNJ-Q2 demonstrated good activity overall (MIC (50/90), 0.015/0.5 μg/mL) and against 3081 S. aureus with > 95% of the isolates inhibited at a MIC of ≤ 0.5 μg/mL; against LVFX-resistant S. aureus isolates, > 90% were inhibited by MIC ≤ 0.5 μg/mL. All isolates were inhibited at a MIC of ≤ 2 μg/mL. JNJ-Q2 have excellent activity (MIC90, 0.015 μg/mL) against isolates of beta-hemolytic streptococci (including Streptococcus pyogenes and S. agalactiae). JNJ-Q2 was the most potent FQL tested overall and against all pathogens when compared directly to MFX, LVFX, and CPFX [17].
The treatment of infections caused by drug-resistant Staphylococcus aureus (DRSA) particularly methicillin-resistant S. aureus (MRSA). Despite the emergence of resistant and MDR S. aureus, seven effective drugs in clinical use for which little resistance has been observed (vancomycin, quinupristin-dalfopristin, telavancin, ceftaroline, linezolid, tigecycline, and daptomycin). However, vancomycin is less effective for infections with MRSA isolates, and has a higher minimum inhibitory concentration (MIC) within the susceptible range. Linezolid is probably the drug of choice for the treatment of complicated MRSA skin and soft tissue infections (SSTIs). Whether drug of choice in pneumonia remains debatable. Daptomycin has shown to be non-inferior to either vancomycin or β-lactams in the treatment of staphylococcal SSTIs, bacteremia, and right-sided endocarditis. Tigecycline was also noninferior to comparator drugs in the treatment of SSTIs, but there is controversy about whether it is less effective than other therapeutic options in the treatment of more serious infections. Telavancin has been shown to be non-inferior to vancomycin in the treatment of SSTIs and pneumonia, but has greater nephrotoxicity. Ceftaroline is a broad-spectrum cephalosporin with activity against MRSA; it is non-inferior to vancomycin in the treatment of SSTIs. Clindamycin, trimethoprim-sulfamethoxazole, doxycycline, rifampin, moxifloxacin (MXF), and minocycline are oral anti-staphylococcal agents that may have utility in the treatment of SSTIs and osteomyelitis, but the clinical data for their efficacy is limited. There are also several drugs with broad-spectrum activity against Gram-positive organisms that have reached the phase II and III stages of clinical testing that will hopefully be approved for clinical use in the upcoming years: oritavancin, dalbavancin, omadacycline, tedizolid, delafloxacin, and JNJ-Q2. Thus, there are currently many effective drugs to treat resistant S. aureus infections and many promising agents in the pipeline. There are still frequent treatment failures and unfortunate clinical outcomes. The clinical challenges presented by MRSA, the clinical experience with current anti-MRSA antibiotics, and the gaps in knowledge on how to use these agents to most effectively combat MRSA infections [11]. JNJ-Q2 is a broad-spectrum FQL with activity against Gram-positive and Gram-negative pathogens. The in vitro activity of JNJ-Q2 was evaluated when tested against Neisseria gonorrhoeae isolates, including ciprofloxacin (CPFX)-resistant strains with documented mutations in the quinolone resistance determining region. MIC values were determined using CPFX, tetracycline, penicillin, ceftriaxone, and azithromycin as comparators. All isolates were inhibited by ≤ 0.25 μg/mL of JNJ-Q2 (range, 0.004-0.25 μg/mL; MIC which was 8-fold (MIC50) and 32-fold more potent (MIC90) compared to CPFX. Few strains were susceptible to penicillin (3.0%) and tetracycline (6.1%), and with the use of the Susceptibility Testing interpretive criteria, 13.6% were resistant to azithromycin. All strains were susceptible to ceftriaxone, the most potent agent (MIC90, 0.06 μg/mL) followed by JNJ-Q2 (MIC90, 0.25 μg/mL). JNJ-Q2 appears to be a potent FQL when tested against contemporary MDR N. gonorrhoeae [12]. JNJ-Q2, a fluorinated 4-quinolone, was very active against both methicillin-susceptible S. aureus (MSSA) and MRSA obtained from patients with acute bacterial skin and skin-structure infection (ABSSSI). Overall MXF and levofloxacin (LVFX) resistance rates were 31.5% and 46.9%, respectively. These favorable results support the continued clinical development of JNJ-Q2 to treat ABSSSI [13]. New antibiotic agents are desperately needed to treat the multidrugresistant (MDR) pathogens that continue to emerge at alarming rates. Many of the agents that have entered full clinical development since 1995 have been members of previously accepted classes of antibiotics. Among these are a new aminoglycoside (plazomicin), anti-MRSA cephalosporins (ceftobiprole, ceftaroline), a monocyclic β-lactam (BAL30072), the β-lactamase inhibitor combination of tazobactam with the anti-pseudomonal cephalosporin ceftolozane, β-lactam combinations with new non-β-lactam inhibitors (MK-7655 with imipenem, and avibactam with ceftazidime and ceftaroline), macrolides (cethromycin and solithromycin), oxazolidinones (tedizolid phosphate and radezolid), and quinolones (delafloxacin, nemonoxacin and JNJ-Q2). Resistance and safety issues have been circumvented by some of these new agents that have well-established mechanisms of action and defined pathways leading toward regulatory approval [14]. JNJ-Q2 is currently in clinical development for the treatment of community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin-structure infections. The JNJ-Q2 showed activity against common respiratory pathogens from patients with CABP. Streptococcus pneumonia (S. pneumoniae), Haemophilus influenzae and Moraxella catarrhalis were tested. JNJ-Q2 had activity against all three species, with 96.9% of strains inhibited at ≤ 0.015 mg/L. JNJ-Q2 [MIC for 50% and 90% of the organisms, respectively demonstrated a 16-fold greater potency compared with MFX and at least 128-fold greater activity compared with levofloxacin (LVFX) and CPFX against S. pneumoniae. The H. influenzae isolates were 21.9-23.3% resistant to ampicillin, but JNJ-Q2 was at least two-fold more active than MXF as well as being potent against M. catarrhalis. The JNJ-Q2 demonstrated increased potency compared with other marketed FQLs that have been used to treat CABP pathogens, thus favouring further clinical development [15]. JNJ-Q2, with anti-MRSA activity, was evaluated in a comprehensive set of non-clinical and clinical cardiovascular safety studies. The effect of JNJ-Q2 on different cardiovascular parameters was compared with that of MFX, sparfloxacin (SPFX) and ofloxacin. Through comparisons with these well-known FQLs, the importance of effects on compensatory ion channels to the cardiovascular safety of JNJ-Q2 was investigated. The trend for effects of JNJ-Q2 on Tp-Te, QT, QRS and PR intervals in the non-clinical models and the plateau in QTc with increasing plasma concentration in humans are consistent with offsetting sodium and calcium channel activities. Based on the non-clinical and clinical cardiovascular safety assessment, JNJ-Q2 has a safe cardiovascular profile for administration in humans with comparable or reduced potential to prolong QT intervals, compared with MFX. The importance of compensatory sodium and calcium channel activity in offsetting potassium channel activity for compounds with a FQL core [16]. JNJ-Q2 have a balanced potency against both DNA gyrase and topoisomerase IV targets. JNJ-Q2 is in the treatment of acute bacterial skin and skin-structure infections (ABSSSIs) and community-acquired bacterial pneumonia. MIC values were obtained for pathogens (44.4% were from patients diagnosed with ABSSSI). JNJ-Q2 demonstrated good activity overall (MIC (50/90), 0.015/0.5 μg/mL) and against 3081 S. aureus with > 95% of the isolates inhibited at a MIC of ≤ 0.5 μg/mL; against LVFX-resistant S. aureus isolates, > 90% were inhibited by MIC ≤ 0.5 μg/mL. All isolates were inhibited at a MIC of ≤ 2 μg/mL. JNJ-Q2 have excellent activity (MIC90, 0.015 μg/mL) against isolates of beta-hemolytic streptococci (including Streptococcus pyogenes and S. agalactiae). JNJ-Q2 was the most potent FQL tested overall and against all pathogens when compared directly to MFX, LVFX, and CPFX [17].
A Mini Review on the Study of New Broad-Spectrum Antimicrobial Fluoroquinolone JNJ-Q2
Mohammad Asif
- Department of Pharmacy, GRD (PG) Institute of Management & Technology, Dehradun, (Uttarakhand), 248009, India
*Address for Correspondence: Mohammad Asif, Department of Pharmacy, GRD (PG) Institute of Management & Technology, Dehradun, Uttarakhand, 248009, India, E-mail: aasif321@gmail.com
Citation: Asif M. A Mini Review on the Study of New Broad-Spectrum Antimicrobial Fluoroquinolone JNJ-Q2. J Chem Applications. 2014;1(1): 5.
Copyright © 2014 Asif M. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Chemistry & Applications | ISSN 2380-5021 | Volume: 1, Issue: 1
Submission: 03 April 2014 | Accepted: 29 May 2014 | Published: 04 June 2014
Abstract
The prokaryotic type II topoisomerases (DNA gyrase and topoisomerase IV) and the eukaryotic type II topoisomerases represent the cellular targets for quinolone antibacterial and anticancer agents. Both enzymes effort the selectively shift from an antibacterial to an antitumor activity. In the search for potential molecule in the quinolones series, some of quinolone analogs displayed antibacterial and cytotoxic activities. The newly developed quinolone JNJ-Q2 is a broad-spectrum fluoroquinolone, has activity against pathogenic bacteria but its anticancer activity is still disclosed. The studies demonstrated that quinolone series changes the biological profile from antibacterial to cytotoxic activity and have excellent potential as antimicrobial and cytotoxic agents.Keywords
Antimicrobial; Anticancer; Fluoroquinolone; New drug;Introduction
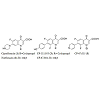
Quinolones are a very important family of antibacterial agents that are widely prescribed for the treatment of infections in humans [1]. They damage the activities of prokaryotic type II topoisomerases, DNA gyrase and topoisomerase IV, and induce them to kill cells by generating high levels of double-stranded DNA breaks. Type II topoisomerases modulate the topological state of the genetic material by passing an intact DNA helix through a transient double stranded break that they generate in a separate DNA segment [2]. Like bacterial cells, eukaryotic species require a type II topoisomerase, known as topoisomerase II, for viability [3]. In addition to the antibacterial quinolones, some quinolones displayed activity against eukaryotic type II topoisomerases, cultured mammalian cells and in vivo tumor models [4]. These antineoplastic quinolones are potentially important as anticancer agents. Several quinolones have been displayed significant activity against eukaryotic type II topoisomerases [5,6]. Quinolones which are built on the ciprofloxacin (CPFX) 1 or norfloxacin (NRFX) 2 nucleus, these selected cytotoxic quinolones 3-5, displayed activity against DNA gyrase or topoisomerase IV. The quinolones are distinguished from the antibacterial quinolones [7,8] and are preferentially target the different prokaryotic and eukaryotic type II topoisomerases [9] (Figure 1).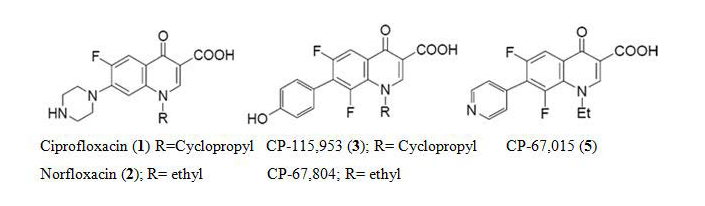
Figure 1: Structures of compounds 1-5.
"The quinolones are divided into generations based on their antibacterial spectrum. The earlier-generation agents are more narrow-spectrum than the later ones. The only standard applied is the grouping of the non-fluorinated drugs found within this quinolones within the ‘first-generation’ heading. The first generation is rarely used today. A number of the second-, third-, and fourth-generation drugs have been removed from clinical practice due to severe toxicity issues. The drugs most frequently prescribed today consist of moxifloxacin, ciprofloxacin, levofloxacin and, to some extent, their generic equivalents (Figure 2, Figure 3).
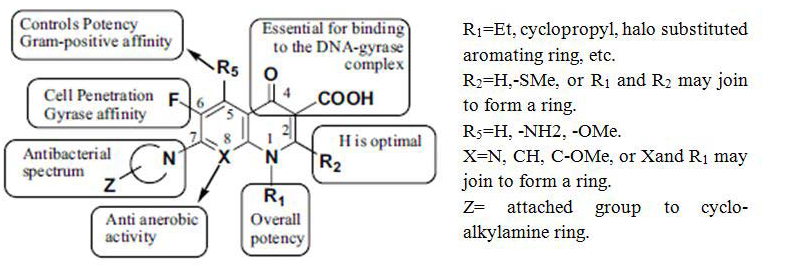
Figure 2: Common structure feature of quinolones.
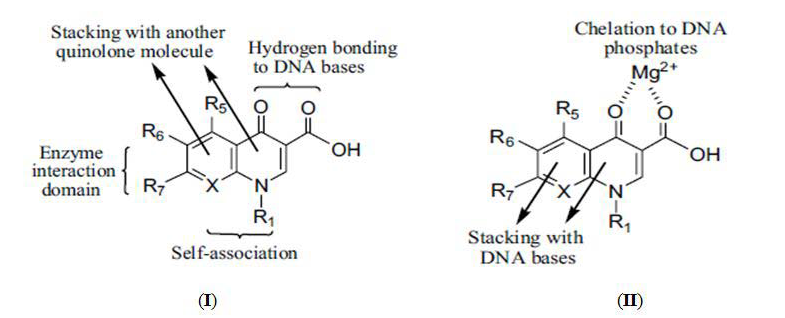
Figure 3: Two binding models (I and II) of quinolones.
Various first generation quinolones such as cinoxacin, flumequine (veterinary use), nalidixic acid, oxolinic acid, piromidic acid, pipemidic acid, and rosoxacin.
Second-generation
The second-generation are ciprofloxacin, enoxacin, fleroxacin, lomefloxacin, nadifloxacin, norfloxacin, ofloxacin, pefloxacin and rufloxacin.
Third-generation
Unlike the first- and second-generations, the third-generation is active against streptococci. Examples are balofloxacin, grepafloxacin, levofloxacin, pazufloxacin, sparfloxacin, temafloxacin, and tosufloxacin.
Fourth-generation
Fourth-generation fluoroquinolones act at DNA gyrase and topoisomerase IV. This dual action slows development of resistance. This dual action slows development of resistance. Examples are clinafloxacin, gatifloxacin, gemifloxacin, moxifloxacin, sitafloxacin, trovafloxacin and prulifloxacin.
In development
Delafloxacin (an anionic fluoroquinoline in clinical trials), JNJ-Q2 (completed Phase II for MRSA), nemonoxacin.
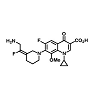
There is an essential need for searching for the new compounds effective in the treatment of infections caused by multidrug-resistant (MDR) bacteria. A series associated with the exploration of new antibacterial agents and it discusses the compounds belonging to the group of quinolones and substances possessing a hybrid structure composed of the quinolone molecule and other compounds. The non-fluorinated quinolone (nemonoxacin, ozenoxacin and KRP-AM 1977X), 6-fluoroquinolones (zabofloxacin, finafloxacin, delafloxacin, JNJ-Q2 (Figure 4), WCK771 and KPI-10), compounds possess a hybrid construction composed of the quinolone structure and other molecules (cadazolid and CBR-2092). These compounds can extend the range of existing antibacterial drugs and provide an alternative to currently available medicinal products [10].
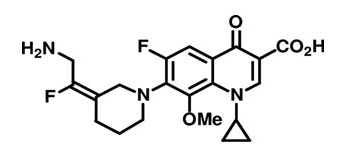
Figure 4: Structures of JNJ-Q2.
A phase II non inferiority study treated patients for 7 to 14 days, testing the efficacy of JNJ-Q2 (250 mg, twice a day [BID]) versus linezolid (600 mg, BID) in patients with acute bacterial skin and skin structure infections (ABSSSI). The prespecified criterion for non inferiority was 15%. Prespecified clinical cure rates 2 to 14 days after completion of therapy were similar (83.1% for JNJ-Q2 versus 82.1% for linezolid). JNJ-Q2 was noninferior to linezolid (61.4% versus 57.7%, respectively). Nausea and vomiting were the most common adverse events. Of the patients, 86% (104/121) had S. aureus isolated from the infection site; 63% of these were MRSA. JNJ-Q2 shows promise as an effective treatment for ABSSSI, demonstrating (i) efficacy for early clinical response, and (ii) cure rates for ABSSSI pathogens (especially MRSA) [18,19]. The in vivo efficacy of JNJ-Q2, was evaluated in a murine septicemia model with MSSA and MRSA and in a S. pneumoniae lower respiratory tract infection model. JNJ-Q2 and comparators were also evaluated in an acute murine skin infection model using a community-acquired MRSA strain and in an established skin infection (ESI) model using a hospital-acquired strain. JNJ-Q2 demonstrated activity in the MSSA septicemia model that was comparable to that MFX (JNJ-Q2 50% effective dose (ED50), 0.2 mg/kg of body weight administered subcutaneously [s.c.] and 2 mg/kg administered orally [p.o.]) and activity in the MRSA septicemia model that was superior to that of vancomycin (JNJ-Q2; ED50), 1.6 mg/kg administered s.c.). In an S. pneumoniae lower respiratory tract infection model, JNJ-Q2 displayed activity (ED50), 1.9 mg/kg administered s.c. and 7.4 mg/kg administered p.o.) that was comparable to that of gemifloxacin and superior to that of MFX. In both MRSA skin infection models, treatment with JNJ-Q2 resulted in dose-dependent reductions in bacterial titers in the skin, with the response to JNJ-Q2 at each dose exceeding the responses of the comparators CPFX, MFX, linezolid, and vancomycin. Additionally, in the ESI model, JNJ-Q2 showed a low or nondetectable propensity for CPFX resistance selection, in contrast to the selection of CPFXresistant mutants observed for both CPFX and MFX. JNJ-Q2 demonstrated activity that was comparable or superior to the activity of FQL or antistaphylococcal comparators in several local and systemic skin infection models performed with both S. aureus and S. pneumoniae [20].
The new broad-spectrum FQL JNJ-Q2 displays in vitro activity against Gram-negative and Gram-positive organisms, including MRSA and CPFX-resistant MRSA isolates. Tested with isogenic MSSA and MRSA strains bearing quinolone-resistant (QR) target mutations, JNJ-Q2 displayed MICs ≤ 0.12 μg/ml, values 16- to 32-fold lower than those determined for MFX. Over expression of the NorA efflux pump did not impact JNJ-Q2 MICs. Inhibition of S. aureus DNA gyrase and DNA topoisomerase IV enzymes demonstrated that JNJ-Q2 was more potent than comparators against wild-type enzymes and enzymes carrying QR amino acid substitutions, and JNJ-Q2 displayed equipotent activity against both enzymes. In serialpassage studies comparing resistance selection in parallel MRSA cultures by CPFX and JNJ-Q2, CPFX readily selected for mutants displaying MIC values of 128 to 512 μg/ml. In contrast, cultures passaged in the presence of JNJ-Q2 displayed MICs ≤ 1 μg/ml for a minimum of 27 days of serial passage. A mutant displaying a JNJ-Q2 MIC of 4 μg/ml was not observed until after 33 days of passage. Mutant characterization revealed that CPFX-passaged cultures with MICs of 256 to 512 μg/ml carried only 2 or 3 quinolone resistance determining region (QRDR) mutations. Cultures passaged with JNJ-Q2 selection for up to 51 days displayed MICs of 1 to 64 μg/ml and carried between 4 and 9 target mutations. CPFX-resistant MRSA exposed to JNJ-Q2 displayed greater decreases in bacterial counts than biofilms exposed to CPFX, MFX, rifampin, or vancomycin [21]. The establish ranges for control strains: S. aureus (0.004 -0.015 μg/ml), Enterococcus faecalis (0.015-0.06 μg/ml), Pseudomonas aeruginosa (0.5 to 2 μg/ml), Escherichia coli (0.008 to 0.03 μg/ml), Haemophilus influenzae (0.002 to 0.015 μg/ml), S. pneumoniae (0.004 to 0.015 μg/ml), and S. aureus. These ranges will be crucial in evaluating JNJ-Q2 potency as it progresses through clinical trial development [22]. The in vitro activity of JNJ-Q2 was evaluated against selected S. aureus samples from patients with acute bacterial skin and skin structure infections. JNJ-Q2 was the most potent FQL tested overall (MIC50 and MIC90, 0.12 and 0.5 μg/ml, respectively) and against methicillin and FQL-resistant subgroups in direct comparisons to MFX, LVFX, and CPFX (each being ≥ 16-fold less potent than JNJ-Q2) [23]. JNJ-Q2, was evaluated for its antibacterial potency and focused on skin and respiratory tract pathogens, including strains exhibiting contemporary FQL resistance phenotypes. Against a set of clinical isolates of S. pneumoniae, including FQL-resistant variants bearing multiple DNA topoisomerase target mutations, an MIC90 value for JNJ-Q2 of 0.12 μg/ml was determined, indicating that it was 32-fold more potent than moxifloxacin (MFX). Against a recently collected MRSA isolates, including 256 CPFX-resistant strains, the JNJ-Q2 MIC90 value was 0.25 μg/ml, similarly indicating that it was 32-fold more potent than MFX. The activities of JNJ-Q2 against Gramnegative pathogens were generally comparable to those of MFX. In further studies, JNJ-Q2 exhibited bactericidal activities against clinical isolates of S. pneumoniae and MRSA with various FQL susceptibilities, and its activities were enhanced over those of MFX. The activity exhibited against strains bearing gyrA, parC, or gyrA plus parC mutations was indicative of the relatively balanced activity of JNJ-Q2 against the DNA topoisomerase target enzymes. The relative rates or frequencies of the spontaneous development of resistance to JNJ-Q2 at MICs in S. pneumoniae, MRSA, and E. coli were indicative of a lower potential for resistance development than that for current FQLs. In conclusion, JNJ-Q2 exhibits a range of antibacterial activities in vitro that is supportive of its further evaluation as a potential new agent for the treatment of skin and respiratory tract infections [24].
Discussion
Although the antibacterial properties and cytotoxic activity of compounds with quinolone pharmacophore are related to their inhibitory activity against topoisomerases. The alteration of biological activity profile of quinolones may be the result of a permeability mechanism or due to the change of selectivity to target enzyme. It appears that the outer membrane of bacteria is the major permeability barrier for quinolones to access their target site and to develop their antibacterial activity [25,26], while quinolones may diffuse directly across the cytoplasmic membrane of tumor cells. The quinolones are relatively simple in structure, mechanistically they are quite complex. The fact that quinolones bind preferentially to enzyme-DNA complexes suggests that quinolones entered the enzyme-drug-DNA ternary complex through interactions with the enzyme-DNA complex, rather than through an association with free nucleic acids. The mode of action of quinolones involves interaction with both prokaryotic and eukaryotic type II topoisomerases. Although the structural features responsible for the interaction of quinolones with the binding sites on prokaryotic or eukaryotic type II topoisomerases are not yet understood fully, position-7 of quinolone structure is considered to be one that directly interacts with topoisomerase enzyme in enzyme-drug-DNA ternary complex, and determines target preference of quinolones [27,10]. Further investigation is required to clarify the action mechanism of compound and understanding the ability of these quinolones to preferentially target the different prokaryotic and eukaryotic type II topoisomerases.References
- Piddock LJV (1999) Mechanisms of fluoroquinolone resistance: An update 1994-1998. Drugs 58: 11-18.
- Drlica K (1999) Mechanism of fluoroquinolone action. Curr Opin Microbiol 2: 504-508.
- Burden DA, Osheroff N (1998) Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochem Biophys Acta 1400: 139-154.
- Clement JJ, Burres N, Jarvis K, Chu DT, Swiniarski J, et al. (1995) Biological characterization of a novel antitumor quinolone. Cancer Res 55: 830-835.
- Anderson VE, Osheroff N (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr Pharm Design 7: 337-353.
- Sissi C, Palumbo M (2003) The quinolone family: from antibacterial to anticancer agents. Curr Med Chem Anti-Cancer Agents 3: 439-450.
- Robinson MJ, Martin BA, Gootz TD, McGuirk PR, Osheroff N (1992) Effects of novel fluoroquinolones on the catalytic activities of eukaryotic topoisomerase II: Influence of the C-8 fluorine group. Antimicrob Agents Chemother 36: 751-756.
- Elsea SH, McGuirk PR, Gootz TD, Moynihan M, Osheroff N (1993) Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential. Antimicrob. Agents Chemother 37: 2179-2186.
- Rajabalian S, Foroumadi A, Shafiee A, Emami S (2007) Functionalized N-(2-oxyiminoethyl) piperazinyl quinolones as new cytotoxic agents. J Pharm Pharm Sci 10: 153-158.
- Karpiuk I, Tyski S (2013) Looking for the new preparations for antibacterial therapy III. New antimicrobial agents from the quinolones group in clinical trials. Przegl Epidemiol 67: 455-460, 557-561.
- Anstead GM, Cadena J, Javeri H (2014) Treatment of infections due to resistant Staphylococcus aureus. Methods Mol Biol 1085: 259-309.
- Biedenbach DJ, Farrell DJ, Flamm RK, Liverman LC, McIntyre G, et al. (2012) Activity of JNJ-Q2, a new fluoroquinolone, tested against contemporary pathogens isolated from patients with community-acquired bacterial pneumonia. Int J Antimicrob Agents 39: 321-325.
- Farrell DJ, Turner LL, Castanheira M, Jones RN (2012) Activity of JNJ-Q2 against Staphylococcus aureus isolated from patients with acute bacterial skin and skin-structure infection obtained during a Phase 2 clinical trial. Diagn Microbiol Infect Dis 74: 73-74.
- Bush K (2012) Improving known classes of antibiotics: an optimistic approach for the future. Curr Opin Pharmacol 12: 527-534.
- Biedenbach DJ, Turner LL, Jones RN, Farrell DJ (2012) Activity of JNJ-Q2, a novel fluoroquinolone, tested against Neisseria gonorrhoeae, including ciprofloxacin-resistant strains. Diagn Microbiol Infect Dis 74: 204-206.
- Eichenbaum G, Pugsley MK, Gallacher DJ, Towart R, McIntyre G, et al. (2012) Role of mixed ion channel effects in the cardiovascular safety assessment of the novel anti-MRSA fluoroquinolone JNJ-Q2. Br J Pharmacol 166: 1694-1707.
- Farrell DJ, Liverman LC, Biedenbach DJ, Flamm RK, Jones RN (2011) Surveillance of JNJ-Q2 activity tested against Staphylococcus aureus and beta-hemolytic streptococci as a component of the 2010 SENTRY Antimicrobial Surveillance Program. Diagn Microbiol Infect Dis 71: 415-420.
- Covington P, Davenport JM, Andrae D, O’Riordan W , Liverman L, et al. (2011) Randomized, double-blind, phase II, multicenter study evaluating the safety/tolerability and efficacy of JNJ-Q2, a novel fluoroquinolone, compared with linezolid for treatment of acute bacterial skin and skin structure infection. Antimicrob Agents Chemother 55: 5790-5797.
- Covington PS, Davenport JM, Andrae DA, Stryjewski ME, Turner LL, et al. (2013) A Phase 2 study of the novel fluoroquinolone JNJ-Q2 in communityacquired bacterial pneumonia. J Antimicrob Chemother 68: 2691-2693.
- Fernandez J, Hilliard JJ, Morrow BJ, Melton JL, Flamm RK, et al. (2011) Efficacy of a new fluoroquinolone, JNJ-Q2, in murine models of Staphylococcus aureus and Streptococcus pneumoniae skin, respiratory, and systemic infections. Antimicrob Agents Chemother 55: 5522-5528.
- Morrow BJ, He W, Amsler KM, Foleno BD, Macielag MJ, et al. (2010) In vitro antibacterial activities of JNJ-Q2, a new broad-spectrum fluoroquinolone. Antimicrob Agents Chemother 54: 1955-1964.
- Ross JE, Liverman LC, Jones RN (2011) Determination of disk diffusion and MIC quality control guidelines for JNJ-Q2, a novel quinolone. J Clin Microbiol 49: 3009-3011.
- Farrell DJ, Liverman LC, Biedenbach DJ, Jones RN (2011) JNJ-Q2, a new fluoroquinolone with potent in vitro activity against Staphylococcus aureus, including methicillin- and fluoroquinolone-resistant strains. Antimicrob Agents Chemother 55: 3631-3634.
- Morrow BJ, Abbanat D, Baum EZ, Crespo-Carbone SM, Davies TA, et al. (2011) Antistaphylococcal activities of the new fluoroquinolone JNJ-Q2. Antimicrob Agents Chemother 55: 5512-5521.
- Nikaido H, Vaara M (1985) Molecular basis of bacterial outer membrane permeability. Microbiol Rev 49: 1-32.
- Neves P, Berkane E, Gameiro P, Winterhalter M, de Castro B (2005) Interaction between quinolones antibiotics and bacterial outer membrane porin OmpF. Biophys Chem 113: 123-128.
- Alovero F, Pan XS, Morris JE, Manzo RH, Fisher LM (2000) Engineering the specificity of antibacterial fluoroquinolones: benzenesulfonamide modifications at C-7 of ciprofloxacin change its primary target in Streptococcus pneumoniae from topoisomerase IV to gyrase. Antimicrob Agents Chemother 44: 320-325.