
Journal of Cardiobiology
Download PDF
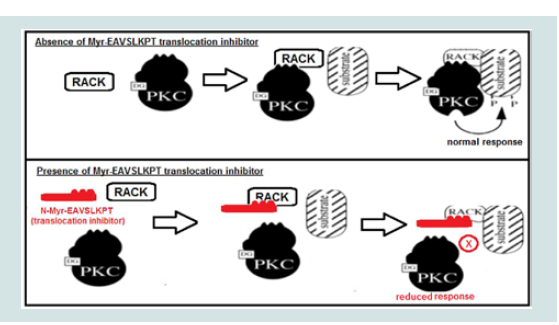 Figure 1: Mechanism of action of myristoylated (Myr)-PKCε- (shown in red, adapted from [36]). PKCε when activated by diacylglycerol (DG) binds to a specific receptor for activated C kinase (RACK) domain which translocates PKCε to interact with substrates (e.g. uncoupled eNOS) to produce a physiologic response (top panel). Myr-PKCε- inhibits PKCε translocation by binding to RACK to reduce the physiologic response evoked by PKCε phosphorylation of its target substrate (bottom panel).
Figure 1: Mechanism of action of myristoylated (Myr)-PKCε- (shown in red, adapted from [36]). PKCε when activated by diacylglycerol (DG) binds to a specific receptor for activated C kinase (RACK) domain which translocates PKCε to interact with substrates (e.g. uncoupled eNOS) to produce a physiologic response (top panel). Myr-PKCε- inhibits PKCε translocation by binding to RACK to reduce the physiologic response evoked by PKCε phosphorylation of its target substrate (bottom panel). 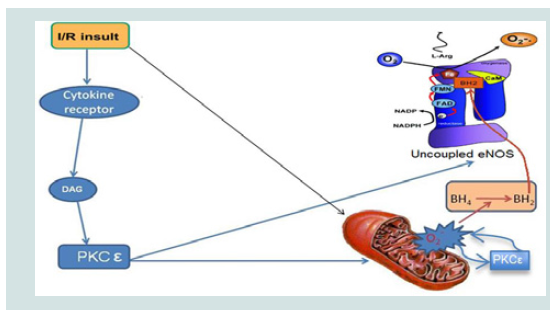 Figure 2: Schematic representation of PKCε regulation of mitochondrial and uncoupled eNOS superoxide (O2-.) release in myocardial I/R (adapted from [37]). I/R insult activates PKC (e.g. PKCε) and causes overproduction of O2-. from damaged mitochondria, thereby leading to oxidation of tetrahydrobiopterin (BH4) to dihydrobiopterin (BH2). BH2 promotes eNOS uncoupling which produces O2-. instead of NO resulting in additional I/R induced oxidative stress. Activated PKCε phosphorylation increases uncoupled eNOS activity and opens mitochondrial ATP-dependent potassium channels during reperfusion leading to increased O2-. induced exacerbation of I/R injury [34,38].
Figure 2: Schematic representation of PKCε regulation of mitochondrial and uncoupled eNOS superoxide (O2-.) release in myocardial I/R (adapted from [37]). I/R insult activates PKC (e.g. PKCε) and causes overproduction of O2-. from damaged mitochondria, thereby leading to oxidation of tetrahydrobiopterin (BH4) to dihydrobiopterin (BH2). BH2 promotes eNOS uncoupling which produces O2-. instead of NO resulting in additional I/R induced oxidative stress. Activated PKCε phosphorylation increases uncoupled eNOS activity and opens mitochondrial ATP-dependent potassium channels during reperfusion leading to increased O2-. induced exacerbation of I/R injury [34,38].
Research Article
Protein Kinase C Epsilon PeptideInhibitor Exerts Cardioprotective Effects in Myocardial Ischemia/Reperfusion Injury
Anahi McIntyre, Christine Adekayode, HannaKim, John Woodley, Tejaswi Dittakavi, Matthew Finnegan, Joseph Heron, Kevin Amuquandoh, Ifeanyi James, Israel Benjamin, Stephanie Liu, Kiana Walker, Tameka Dean, Wesley Hwang, Qian Chen, Robert Barsotti and Lindon H. Young*
- Bio-Medical Sciences, Philadelphia College of Osteopathic Medicine, USA
*Address for Correspondence: Lindon H. Young, Bio-Medical Sciences, Philadelphia College ofOsteopathic Medicine, 4170 City Avenue, Philadelphia, PA 19131, USA, Tel: 215-871-6832; E-mail: lindonyo@pcom.edu
Citation: McIntyre A, Adekayode C, Kim H, Woodley J, Dittakavi T, et al. Protein Kinase C Epsilon Peptide Inhibitor Exerts Cardioprotective Effects in Myocardial Ischemia/Reperfusion Injury. J Cardiobiol. 2018;5(1): 6.
Copyright: © 2017 McIntyre A, et al. This is an open access articledistributed under the Creative Commons Attribution License, whichpermits unrestricted use, distribution, and reproduction in any medium,provided the original work is properly cited.
Journal of Cardiobiology | ISSN: 2332-3671 | Volume: 5, Issue: 1
Submission: 23 November, 2017 | Accepted: 08 January, 2018 | Published: 15 January, 2018
Keywords
Myocardial ischemia/reperfusion injury; Protein kinase C epsilon; Endothelial nitric oxide synthase
Abstract
Background: The generation of reactive oxygen species (ROS) during myocardial ischemia (I)/reperfusion (R) contributes to postreperfusion cardiac injury. The increase in ROS is attributed in part to the uncoupling of endothelial nitric oxide synthase (eNOS) that produces ROS instead of nitric oxide (NO) and mitochondrial derived ROS which in part is derived from opening of mitochondrial ATPsensitive K+ channels (mKATP). Both the activity of uncoupled eNOS and the opening of mKATP channels in mitochondria are stimulated by protein kinase C epsilon (PKCε) that is activated during reperfusion. We hypothesize that a cell permeable PKCε peptide inhibitor, (N-myristic acid-EAVSLKPT, Myr-PKCε-) given at reperfusion will improve postreperfused cardiac function and attenuate infarct size compared to untreated isolated perfused rat hearts subjected to ischemic reperfusion injury.
Methods: Male Sprague-Dawley rats (275-325 g) were anesthetized with sodium pentobarbital (60 mg/kg) and anticoagulated with heparin 1000 units given intraperitoneally. The hearts were excised and attached to a Langendorff perfusion system. All hearts were subjected to 30 min of global ischemia, followed by 90 min of reperfusion. Myr- PKCε- was dissolved in Krebs’ buffer and infused during the first 10 min of reperfusion at final concentrations of 5, 10 and 20 μM.
Results: Myr-PKCε- treated hearts (10 and 20 μM) exhibited significant improvement in post-reperfused cardiac function at 90 min compared to untreated controls. The maximal rate of left ventricular developed pressure generation (+dP/dtmax) of Myr-PKCε- treated hearts showed significant recovery to 56±4% (10 μM, p<0.05, n=8) and 50±3%; (20 μM, p<0.05, n=8) of initial baseline values. By contrast, the +dP/ dtmax of both untreated control hearts (n=9) and those treated with the lowest concentration of Myr-PKCε- (5 μM, n=8) recovered to only 30±4% and 33±4% of initial baseline values, respectively. Interestingly, all Myr-PKCε- treated hearts (5-20 μM) showed significant reduction in infarct size to 28±2% compared to untreated control hearts, which averaged 38±3% (p<0.05, n=9).
Conclusion: The results suggest that Myr-PKCε- given at reperfusioneffectively reduces infarct size, improves cardiac function and is aputative treatment that could aid in clinical myocardial infarction/organ transplantation patient recovery.
Introduction
Myocardial cell death from ischemia-reperfusion (I/R) injury (e.g. myocardial infarction) continues to be a major cause of morbidity and mortality in Western nations [1]. Reperfusion is essential tosalvage ischemic myocardial tissue from infarction. However, there-establishment of blood flow to acutely ischemic myocardium leads to rapid cell death and compromised heart function [2]. The size of the infarct that results from the combination of the ischemic and reperfusion injury is the major determinant of the prognosis of patients who survive the acute myocardial infarction incident [3-7]. The necessity for effective pharmacological intervention still exists, despite numerous basic studies firmly establishing that I/R injury can be delayed or even reduced by the introduction of cardioprotective agents or strategies prior to a prolonged ischemic insult (preconditioning) or at the start of reperfusion [1,8]. Such interventions have been shown to reduce infarct size, attenuate the frequency and severity of reperfusion-induced arrhythmias, and prevent endothelial dysfunction [9-13].
Preconditioning of the myocardium can be induced by several rounds of brief ischemia (i.e. less than 4 min) followed by several rounds of brief reperfusion (i.e. less than 6 min) prior to prolonged ischemia (i.e. 30 min or greater) followed by the final reperfusion period [12]. The precise mechanism for ischemic preconditioning remains unclear; however, there is growing evidence that activation of protein kinase C epsilon (PKCε) plays a central role [12,14-16]. The transient ischemic episodes are thought to activate PKCε directly by cytokine induced production of intracellular diacylglycerol (DAG) and indirectly by eliciting a small amount of ROS release from the transient opening of the mitochondrial ATP-sensitive K+ channels (mKATP) on the inner mitochondrial membrane. ROS activates PKCε via oxidative modification of its regulatory domain. Activated PKCε translocates to the mKATP channel releasing additional ROS which in turn further activates PKCε in a positive feedback loop [17-19]. The resulting ROS then activates a second PKCε mitochondrial pool that prevents the opening of the mitochondrial permeability transition pore (MPTP). The MPTP is a large conductance channel in the inner mitochondrial membrane whose extended opening leads to the dissipation of the inner mitochondrial membrane potential, matrix swelling and rupture of the outer mitochondrial membrane, ultimately leading to necrosis [20-24].
Results from several studies generally support the role of PKCε activation in preconditioning, for example, agents that mimic preconditioning also activate PKCε such as adenosine, volatile anesthetics, and exogenously applied PKCε agonists [16,25-28]. Moreover, preconditioning of isolated hearts from PKCε knockout mice failed to reduce I/R injury [12]. Conversely, in studies in which PKCε activity was promoted using a PKCε peptide activator (HDAPIGYD) given during reperfusion rather than prior to ischemia, no cardioprotective effects were exhibited [13,29].
Our research group confirmed these findings and discovered that a cell permeable myristic acid conjugated PKCε peptide inhibitor (Myr-PKCε-) dramatically attenuated cardiac contractile dysfunction in leukocyte-augmented myocardial I/R injury when given during reperfusion [13]. Myr-PKCε- acts as a competitive translocation inhibitor by mimicking the receptor for activated C kinase (RACK) binding site on PKCε (see Figure 1) [30]. During reperfusion in this I/R model, three principle sources of ROS will be reduced directly and indirectly by inhibition of PKCε activity, opening of the mKATP channels, uncoupled endothelial nitric oxide synthase (eNOS)activity, a switch from nitric oxide (NO) to ROS production, and intracellular adhesion molecule-1 (ICAM-1) mediated leukocyte ROS release (see Figure 2) [31-35]. We previously reported that PKCε activation upon reperfusion is not cardioprotective, unless eNOS uncoupling is reversed to its normal or coupled state by the addition of an essential cofactor, tetrahydrobiopterin (BH4) [34]. During reperfusion, eNOS uncoupling is promoted by BH4 oxidation to dihydrobiopterin (BH2). Since BH2 and BH4 have equal affinity forthe eNOS oxygenase-binding domain, an increase in the BH2 to BH4 ratio during reperfusion shifts eNOS towards the uncoupled state [34]. Hence, inhibiting PKCε during reperfusion will attenuate ROS mediated insults arising from uncoupled eNOS activity, the mKATP channel, and leukocyte endothelial adhesion and penetration.
We previously found that Myr-PKCε- (10 μM) given at reperfusion elicited cardioprotective effects in an isolated perfused rat heart model subjected to I (30 min)/R (45 min) [39]. The aim of the present study is to determine the optimal concentration for the cardioprotective effects of Myr-PKCε- when the reperfusion period is doubled to 90 min.
Methods
Concentration-dependent cardioprotective effects of Myr-PKCε- (5, 10 and 20 μM) were evaluated based on improved cardiac function and reduction of infarction size compared to untreated (control) I/R hearts.
Male Sprague-Dawley rats (275-325 g, Charles River, Springfield, MA) were anesthetized via an intraperitoneal injection of sodium pentobarbital (60 mg/kg) and anticoagulated with heparin 1000 units. Hearts were quickly isolated and attached to a Langendorff apparatus as previously described [39]. The I/R protocol consisted of a 15 min stabilization period, 30 min of global ischemia, and 90 min of reperfusion. The following cardiac function parameters were measured throughout the entire protocol using a data acquisition system (PowerLab/8Sp, ADInstruments, Colorado Springs, CO). Heart rate, left ventricular end systolic pressure (LVESP), enddiastolic pressure (LVEDP), left ventricular developed pressure (LVDP, i.e. LVESP-LVEDP), and the maximal rate of left ventricular pressure generation (+dP/dtmax) and decline (-dP/dtmin) were measured using a pressure transducer (SPR-524; Millar Instruments, Inc., Houston, TX) placed in the left ventricle. Coronary flow was monitored by a flowmeter (T106; Transonic Systems, Inc., Ithaca, NY). A stock of Myr-PKCε- (N-Myr-EAVSLKPT; MW=1054 g/ mol; (Genemed Synthesis, Inc., San Antonio, TX) was prepared by solubilizing the appropriate amount of the compound in 28% dimethyl sulfoxide (DMSO). This stock solution was further diluted to yield a final concentration of 5, 10 or 20 μM in 0.03% DMSO in the perfusate. This compound was then infused during the first 10 min of reperfusion at a rate of 1 ml/min via a syringe pump.
To determine infarct size, all hearts were removed from the apparatus, lightly blotted, and placed in a -20 °C freezer for 30 min. The partially frozen heart was sectioned perpendicular to its long axis into 2 mm thick slices. The slices were then incubated at 37 °C in a 0.2 M Tris buffer (pH=7.41) containing 1% 2,3,5-triphenyltetrazolium chloride (TTC) for ~5 min to demarcate the infarcted (white) from the viable (red) tissue. Stained heart slices were transferred to 4% paraformaldehyde solution for ~15 min to improve the contrast between the infarcted and viable tissue areas. The infarct size was determined by carefully dissecting (under magnification) and weighing the corresponding tissue in both areas. The infarcted tissue weight was divided by total weight to yield percentage of infarcted tissue.
Statistical Analysis 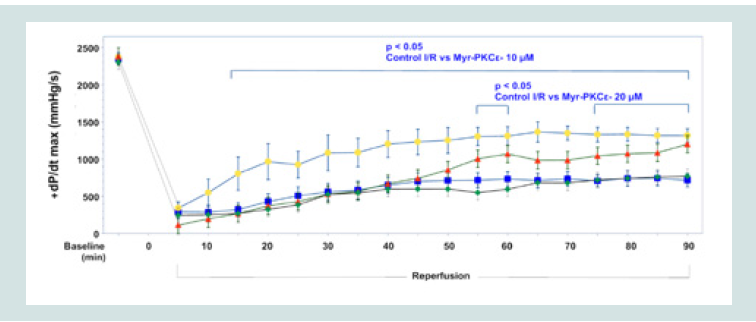 Figure 3: Time course of +dP/dtmax for Control I/R (blue), I/R+Myr-PKCε- 5μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red).
Figure 3: Time course of +dP/dtmax for Control I/R (blue), I/R+Myr-PKCε- 5μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red). 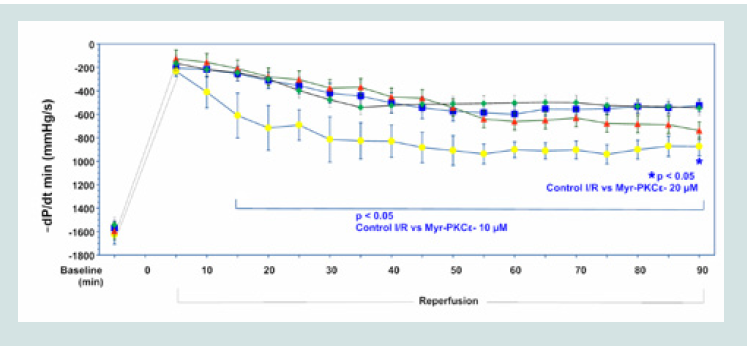 Figure 4: Time course of -dP/dtmin for Control I/R (blue), I/R+Myr-PKCε- 5μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red).
Figure 4: Time course of -dP/dtmin for Control I/R (blue), I/R+Myr-PKCε- 5μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red). 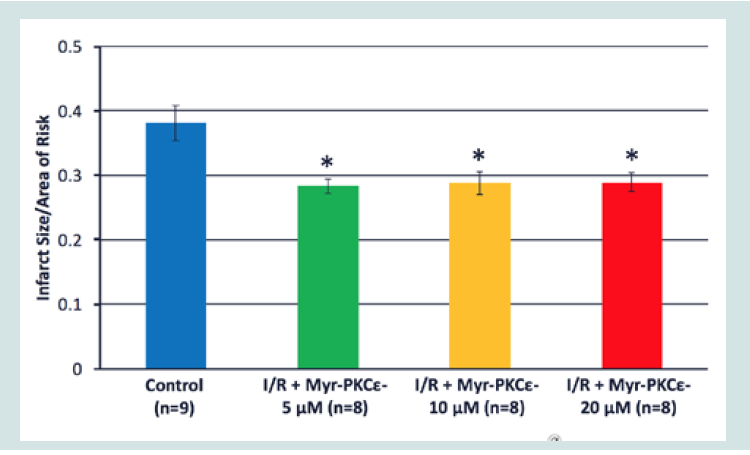 Figure 5: Percent infarcted size of control I/R (blue), I/R+Myr-PKCε- 5 μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red) after TTC staining.*p<0.05 vs. control I/R hearts.
Figure 5: Percent infarcted size of control I/R (blue), I/R+Myr-PKCε- 5 μM (green), I/R+Myr-PKCε- 10 μM (yellow) and I/R+Myr-PKCε- 20 μM (red) after TTC staining.*p<0.05 vs. control I/R hearts. 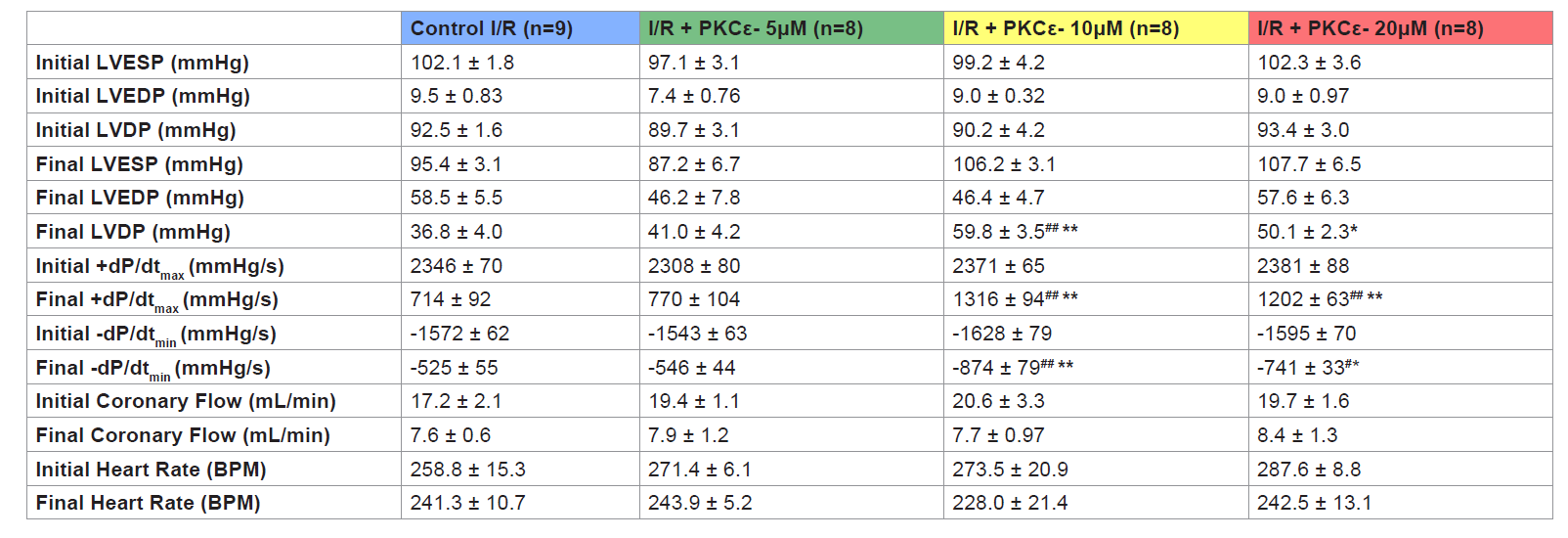 Table 1: Table 1: Cardiac function initial (baseline) and final values for control I/R and I/R+Myr-PKCε- (5, 10 and 20 μM) treated hearts. LVESP, left ventricular end-systolicpressure; LVEDP, left ventricular end-diastolic pressure; LVDP, left ventricular developed pressure; maximal rate of LV pressure generation (+dP/dtmax) and decline (-dP/dtmin). Data expressed as mean±S.E.M.*p < 0.05; **p < 0.01 vs. control I/R. #p < 0.05; ##p < 0.01 vs. I/R + Myr-PKCε- (5 μM).
Table 1: Table 1: Cardiac function initial (baseline) and final values for control I/R and I/R+Myr-PKCε- (5, 10 and 20 μM) treated hearts. LVESP, left ventricular end-systolicpressure; LVEDP, left ventricular end-diastolic pressure; LVDP, left ventricular developed pressure; maximal rate of LV pressure generation (+dP/dtmax) and decline (-dP/dtmin). Data expressed as mean±S.E.M.*p < 0.05; **p < 0.01 vs. control I/R. #p < 0.05; ##p < 0.01 vs. I/R + Myr-PKCε- (5 μM).
All data are presented as the mean±S.E.M. ANOVA analysis using Student-Neuman-Keuls test was used to assess statistical difference in cardiac function parameters and infarct size between control (untreated) I/R and I/R+Myr-PKCε- (treated) hearts. Probability values of <0.05 were considered statistically significant.
Results
Hearts treated at reperfusion with 10 and 20 μM Myr-PKCε-, but not with 5 μM, exhibited improvement in the following parameters of cardiac function following 90 min of reperfusion: LVDP, and both dP/dtmax and min (see Table 1). The higher concentrations of Myr- PKCε- (10 and 20 μM) significantly restored +dP/dtmax to 56±4% (p<0.05, n=8) and 50±3% (p<0.05, n=8) compared to treatment with lower concentrations of Myr-PKCε- (5 μM) and untreated control hearts which only recovered to 33±4% (n=8) and 30±4% (n=9) of initial baseline values respectively at the end of the reperfusion period (Figure 3).
Hearts treated with the lowest concentration of Myr-PKCε- (5 μM, n=8) recovered to 35±3% of baseline -dP/dtmin, which was not significantly different from untreated controls (Figure 4). While hearts treated with higher concentrations of Myr-PKCε- showed significant restoration in left ventricular -dP/dtmin at the end of the reperfusion period to 54±5% with 10 μM (p<0.05, n=8) and to 46±2% with 20 μM (p<0.05, n=8) of initial baseline values compared to untreatedcontrols, which only recovered to 33±3% (n=9), respectively.
Hearts treated with higher concentrations of Myr-PKCε- (10 and 20 μM) exhibited significant improvement in post-reperfused LVDP. Recovery of LVDP reached 66±4% with 10 μM (p<0.01, n=8) and 54±3% with 20 μM (p<0.05, n=8) at 90 min of reperfusion compared to hearts treated with 5 μM (n=8) and control (n=9) hearts which only recovered to 46±5% and 40±4% of initial baseline values at the end of the reperfusion period (Table 1).
At all concentrations, Myr-PKCε- treated hearts showed significant reduction in infarct size, 28±2% (5 μM, n=8), 29±2% (10 μM, n=8) and 29±2% (20 μM, n=8), compared to untreated control I/R hearts, 38±3% (p<0.05, n=9) (Figure 5).
Discussion
Treatment with Myr-PKCε- at the onset of reperfusion was effective in restoring post-reperfused cardiac function at concentrations of 10 and 20 μM (see Figure 3 and 4) (Table 1) and reducing infarct size at all concentrations tested (5, 10 and 20 μM, see Figure 5 ). The time course of the recovery of cardiac function was slightly slower following 20 μM compared to 10 μM Myr-PKCε-. By contrast, 5 μM treated hearts showed no significant improvement in function throughout reperfusion compared to untreated control hearts. Nevertheless, infarct size in Myr-PKCε- treated hearts (5, 10 and 20 μM) was approximately the same: 28% (5 μM) and ~29% (10 and 20 μM). Yet, all treated hearts showed significant reduction in infarct size compared to controls (~38%, p<0.05). Interestingly, in a porcine acute intact regional I (1 hr)/R (3 hr) model, Montgomery M et al. showed that Myr-PKCε- given IV at reperfusion to yield an estimated circulating concentration of 10 μM (0.8 mg/kg) restored post-reperfused cardiac function (i.e. ejection fraction) by 91±3% of initial baseline values and reduced infarct size to 13±0.3% [39]. In comparison, untreated controls only recovered to 69±3% of initial baseline and had infarct sizes of 34±4%. Collectively, the results from these studies indicate that the cardioprotective effects of Myr-PKCε-are consistent across species and models of I/R injury.
It is unclear why the 5 μM treated hearts failed to show significant recovery in function despite a lower infarct size than control hearts. These results suggest that Myr-PKCε- effectively inhibits necrosis independently of function recovery, which may be due to continually elevated ROS levels in the low concentration treated hearts (i.e. 5 μM) that cause stunning. Previous myocardial I/R studies using a ROS-reducing antioxidant peptide targeted to the mitochondria have also reported a reduction in infarct size without improvement in contractility lasting days to weeks after reperfusion [40]. Moderately elevated levels of ROS could potentially contribute to stunning of reversibly damaged myocardial tissue while higher levels lead to necrosis. The lack of a concentration-dependent improvement in infarct size with increasing Myr-PKCε-concentrations indicates that the ED50 for this molecule on infarct size is markedly below 5 μM.
The absence of a concentration dependent effect of a cardioprotective agent on infarct size has been reported in other studies. Arakawa M et al. found no difference in infarct size in isolated perfused rat hearts treated with three concentrations (i.e. 5, 50, and 500 nM) of an anti-apoptotic drug (FNK) constructed from Bcl-x(L), a member of the Bcl-2 apoptotic regulator protein family [41]. In their study, all three concentrations tested resulted in a reduction in infarct size to ~30±5% (n=8) compared to 47±5% (n=8) in control hearts. Similar to our current study, the middle concentration tested (i.e.50 nM) restored post-reperfused cardiac function (e.g. LVDP) more so than the higher and lower concentrations and was significantly different from controls. In another study of isolated perfused rat hearts, Abarbanell AM et al. reported no difference in infarct size between high (i.e. 1 μg) and low concentrations (i.e. 200 ng; ~38±2%, n=10) of a cytokine mediated inhibitor (HMGB1) given during reperfusion compared to controls (49±3%, n=9) despite differences in post-reperfused cardiac function (i.e. +dP/dtmax and -dP/dtmin) [42]. The infarct size reduction reported in drug treated hearts (i.e. 23% of control) is similar to our results (i.e. 26% of control). Lastly, Zhang W et al. reported that aliskiren, a direct renin inhibitor, lowered blood pressure in a concentration dependent manner, whereas there was no difference in infarct size in rats subjected to regional cardiac I/R between the high (i.e. 60 mg/kg, lower blood pressure) and low concentrations (i.e. 30 mg/kg) of aliskiren [43]. Collectively, the results from these three studies targeting different cellular pathways are comparable to our current findings and suggest that there may be a finite efficaciousness to pharmacological interventions targeted at reducing infarct size when given during reperfusion. The primary mechanism that underlies this early lethal reperfusion injury is still unknown but likely includes Ca2+ dysregulation and changes in pH that occur during the ischemic phase. The influx of Ca2+ and the rapid restoration of physiologic pH during reperfusion leads to rapid dysfunction, hypercontracture and necrosis [44]. Thus deep reductions in infarct size during I/R injury may only be possible with preconditioning treatment that acts to limit and/or correct for this Ca2+ and pH dysregulation which begins during the ischemic phase[44].
The time course of recovery in function of +dP/dtmax and -dP/dtminappeared to be optimal in the hearts treated with 10 μM Myr-PKCε-. By comparison, we previously showed that 10 μM concentration significantly restored cardiac function (i.e. +dP/dtmax) at 10 min reperfusion and maintained this effect throughout the reperfusion period whether it was 45 or 90 min reperfusion [39]. The results for the current study clearly shows that the 10 μM concentration PKCε- can maintain its cardioprotective effect for an additional 45 min without significantly altering infarct size [i.e. 25±2% (45 min reperfusion) vs. 29±2% (90 min reperfusion)] [39]. The relatively slow recovery of function in the 20 μM treated hearts suggests that this higher concentration may cause some direct stunning by the drug that in turn slows the rate of recovery. Similarly, Abarbanell AM et al. showed that higher concentration of a cytokine inhibitor did not result in improved cardiac function [42]. Interestingly, Koyanagi T et al. reported that PKCε- inhibits cytokine production after heart transplantation [45]. Therefore, it is possible that the higher drug concentrations may stun the myocardium slowing the rate of functional recovery. By contrast, hearts subjected to the lower concentration of Myr-PKCε- (5 μM) may reduce ROS production to a level that attenuates infarct size, but not sufficient to improve post-reperfused cardiac function (see LVDP, +dP/dtmax and -dP/ dtmin data in Table 1). We surmise that the salvaged cardiomyocytes would exhibit improved contractile function after stunning subsides, which could occur days to weeks after ischemic insult in vivo [40]. Further chronic myocardial I/R studies using Myr-PKCε- (5 μM) are needed in vivo to determine if stunning subsides after treatment. In toto, these results suggest that the 10 μM concentration was optimal to restore post-reperfused cardiac function and reduce infarct size.
We previously reported that 5 μM Myr-PKCε- resulted in 87% recovery of +dP/dtmax initial baseline values using an I/R model in which isolated perfused rat hearts were subjected to 20 min of global ischemia followed by 5 min of reperfusion with a Krebs’ solution containing 2 x 108 leukocytes and 40 min of leukocyte-free buffer [13]. By contrast, in the present study at the same PKCε- concentration (5 μM) no significant recovery of +dP/dtmax and -dP/dtmin compared to controls occurred. Infarct size was not measured in the previous study. We speculate that in a leukocyte augmented I/R model damage as evidenced by an impairment in function was synergistically produced by the leukocyte infusion and the 20 min ischemic period. Myr-PKCε- treatment possibly reduced leukocyte vascular adhesion and diapedesis into the myocardium, lowering leukocyte ROS release, and resulting in less cardiac dysfunction. Thus the almost complete recovery of post-reperfused cardiac function was due to blocking the leukocyte-mediated component of the damage. By contrast, the current study employed a longer 30 min ischemic period and no leukocytes. Nevertheless, in both I/R models, Myr-PKCε- given at reperfusion markedly improved post-reperfused cardiac function (i.e. +dP/dtmax and -dP/dtmin). A putative mechanism for this effect in either I/R model is that treatment with Myr-PKCε- inhibits uncoupled eNOS activity, resulting in an increase in the bioavailability of NO and inhibition of degradation of NO produced from other sources such as hemoglobin and myoglobin nitrite reductase [13,34]. NO has been shown to promote a number of beneficial actions during normal cardiac activity and I/R injury, such as reduction in O2 consumption, inhibition of Ca2+ influx, and improvement in the coupling efficiency between ATP synthesis and O2 consumption [46-50]. In addition, constitutive NO stimulation of Na+/K+-ATPase has been reported to protect against mitochondrial membrane collapse and inhibits the formation of MPTP during reperfusion [51]. Collectively, these NO effects would improve cardiac function and reduce cell death (infarct size) during reperfusion. Interestingly, aliskiren is purported to increase NO bioavailability via increased eNOS phosphorylation when given prior to prolonged myocardial ischemia. This effect was blocked by NG-nitro-L-arginine methyl ester [43]. It is possible that a threshold of NO bioavailability is obtained at the lower concentration (i.e. 5 μM) that is sufficient to limit infarct size independently from the effects of increasing concentrations of Myr-PKCε- that would improve post-reperfused cardiac function.
Several other animal studies have shown that chronic treatment with a cell permeable PKCε- was safe and effective in attenuating both cardiac and vascular remodeling following transplantation and angioplasty [45,52]. The purported mechanism mediating these chronic PKCε- effects was the inhibition of cytokine-induced metalloproteinase expression and subsequent associated extracellular fibrosis [45,53]. Consequently, acute administration of cell permeable PKCε- given at the onset of reperfusion may be a novel clinical therapy to attenuate cardiac contractile dysfunction and cell death following a heart attack, while chronic administration could be used to attenuate tissue remodeling following cardiac balloon angioplasty/stenting and transplantation.
Conclusion
Collectively, the results show that Myr-PKCε- treatment at the onset of reperfusion reduced infarct size at all concentrations tested while post-reperfused cardiac function markedly recovered at the higher concentrations. Thus, the data suggest that Myr- PKCε- treatment would be an effective clinical strategy to attenuate myocardial reperfusion injury in heart attack and transplant patients.
References
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. (2015) Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 131: e29-e322.
- Bulluck H, Yellon DM, Hausenloy DJ (2016) Reducing myocardial infarct size: challenges and future opportunities. Heart 102: 341-348.
- Lassen JF, Botker HE, Terkelsen CJ (2013) Timely and optimal treatment of patients with STEMI. Nat Rev Cardiol 10: 41-48.
- Lonborg J, Engstrom T (2013) Correlating infarct size and patient prognosis: are cardiac biomarkers truly insufficient? Eur Heart J Cardiovasc Imaging 14: 932-933.
- Burns RJ, Gibbons RJ, Yi Q, Roberts RS, Miller TD, et al. (2002) The relationships of left ventricular ejection fraction, end-systolic volume index and infarct size to six-month mortality after hospital discharge following myocardial infarction treated by thrombolysis. J Am Coll Cardiol 39: 30-36.
- Ndrepepa G, Kastrati A, Mehilli J, Neumann FJ, ten Berg J, et al. (2008) One-year clinical outcomes with abciximab vs. placebo in patients with non-ST-segment elevation acute coronary syndromes undergoing percutaneous coronary intervention after pre-treatment with clopidogrel: results of the ISAR-REACT 2 randomized trial. Eur Heart J 29: 455-461.
- Kelle S, Roes SD, Klein C, Kokocinski T, de Roos A, et al. (2009) Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J Am Coll Cardiol 54: 1770-1777.
- Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123: 92-100.
- Inagaki K, Begley R, Ikeno F, Mochly-Rosen D (2005) Cardioprotection by epsilon-protein kinase C activation from ischemia: continuous delivery and antiarrhythmic effect of an epsilon-protein kinase C-activating peptide. Circulation 111: 44-50.
- Alkaitis MS, Crabtree MJ (2012) Recoupling the cardiac nitric oxide synthases: tetrahydrobiopterin synthesis and recycling. Curr Heart Fail Rep 9: 200-210.
- Hearse DJ, Tosaki A (1987) Free radicals and reperfusion-induced arrhythmias: protection by spin trap agent PBN in the rat heart. Circ Res 60: 375-383.
- Saurin AT, Pennington DJ, Raat NJ, Latchman DS, Owen MJ, et al. (2002) Targeted disruption of the protein kinase C epsilon gene abolishes the infarct size reduction that follows ischaemic preconditioning of isolated buffer-perfused mouse hearts. Cardiovasc Res 55: 672-680.
- Teng JC, Kay H, Chen Q, Adams JS, Grilli C, et al. (2008) Mechanisms related to the cardioprotective effects of protein kinase C epsilon (PKC epsilon) peptide activator or inhibitor in rat ischemia/reperfusion injury. Naunyn Schmiedebergs Arch Pharmacol 378: 1-15.
- Baines CP, Goto M, Downey JM (1997) Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol 29: 207-216.
- Ping P, Song C, Zhang J, Guo Y, Cao X, et al. (2002) Formation of protein kinase C(epsilon)-Lck signaling modules confers cardioprotection. J Clin Invest 109: 499-507.
- Liu GS, Cohen MV, Mochly-Rosen D, Downey JM (1999) Protein kinase C-epsilon is responsible for the protection of preconditioning in rabbit cardiomyocytes. J Mol Cell Cardiol 31: 1937-1948.
- Gopalakrishna R, Anderson WB (1989) Ca2+- and phospholipid-independent activation of protein kinase C by selective oxidative modification of the regulatory domain. Proc Natl Acad Sci U S A 86: 6758-6762.
- Korichneva I, Hoyos B, Chua R, Levi E, Hammerling U (2002) Zinc release from protein kinase C as the common event during activation by lipid second messenger or reactive oxygen. J Biol Chem 277: 44327-44331.
- Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, et al. (1997) Activation of protein kinase C by tyrosine phosphorylation in response to H2O2. Proc Natl Acad Sci U S A 94: 11233-11237.
- Griffiths EJ, Halestrap AP (1995) Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 307: 93-98.
- Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR (2004) Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 287: H841-H849.
- Hunter DR, Haworth RA, Southard JH (1976) Relationship between configuration, function, and permeability in calcium-treated mitochondria. J Biol Chem 251: 5069-5077.
- Halestrap AP, Clarke SJ, Javadov SA (2004) Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 61: 372-385.
- Korge P, Honda HM, Weiss JN (2002) Protection of cardiac mitochondria by diazoxide and protein kinase C: implications for ischemic preconditioning. Proc Natl Acad Sci U S A 99: 3312-3317.
- Dorn GW 2nd, Souroujon MC, Liron T, Chen CH, Gray MO, et al. (1999) Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci U S A 96: 12798-12803.
- Lancaster TS, Jefferson SJ, Korzick DH (2011) Local delivery of a PKCε-activating peptide limits ischemia reperfusion injury in the aged female rat heart. Am J Physiol Regul Integr Comp Physiol 301: R1242-R1249.
- Maslov LN, Khaliulin I, Oeltgen PR, Naryzhnaya NV, Pei JM, et al. (2016) Prospects for creation of cardioprotective and antiarrhythmic drugs based on opioid receptor agonists. Med Res Rev 36: 871-923.
- Budas GR, Churchill EN, Mochly-Rosen D (2007) Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol Res 55: 523-536.
- Inagaki K, Hahn HS, Dorn GW 2nd, Mochly-Rosen D (2003) Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 108: 869-875.
- Mochly-Rosen D, Gordon AS (1998) Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J 12: 35-42.
- Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD (2006) Mitochondrial PKCε and mitochondrial ATP-sensitive K+ channel copurify and coreconstitute to form a functioning signaling module in proteoliposomes. Circ Res 99: 878-883.
- Ago T, Kuroda J, Kamouchi M, Sadoshima J, Kitazono T (2011) Pathophysiological roles of NADPH oxidase/nox family proteins in the vascular system. -Review and perspective-. Circ J 75: 1791-1800.
- Zhang J, Baines CP, Zong C, Cardwell EM, Wang G, et al. (2005) Functional proteomic analysis of a three-tier PKCepsilon-Akt-eNOS signaling module in cardiac protection. Am J Physiol Heart Circ Physiol 288: H954-H961.
- Perkins KA, Pershad S, Chen Q, McGraw S, Adams JS, et al. (2012) The effects of modulating eNOS activity and coupling in ischemia/reperfusion (I/R). Naunyn Schmiedebergs Arch Pharmacol 385: 27-38.
- Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, et al. (1998) Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98: 699-710.
- Csukai M, Mochly-Rosen D (1999) Pharmacologic modulation of protein kinase C isozymes: the role of RACKs and subcellular localisation. Pharmacol Res 39: 253-259.
- Chen Q, Parker CW, Devine I, Ondrasik R, Habtamu T, et al. (2016) Apocynin exerts dose-dependent cardioprotective effects by attenuating reactive oxygen species in ischemia/reperfusion. Cardiovasc Pharm Open Access 5: 1-7.
- Pomerantz BJ, Robinson TN, Heimbach JK, Calkins CM, Miller SA, et al. (2000) Selective mitochondrial KATP channel opening controls human myocardial preconditioning: Too much of a good thing? Surgery 128: 368-373.
- Montgomery M, Adams J, Teng JC, Tekelehaymanot B, Ondrasik R, et al. (2013) Myristoylated protein kinase C epsilon peptide inhibitor exerts cardioprotective effects in rat and porcine myocardial ischemia/reperfusion: a translational research study. Proceedings of the 23rd American Peptide Symposium, American Peptide Society, USA, pp. 178-179.
- Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, et al. (2012) Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J Am Heart Assoc 1: e001644.
- Arakawa M, Yasutake M, Miyamoto M, Takano T, Asoh S, et al. (2007) Transduction of anti-cell death protein FNK protects isolated rat hearts from myocardial infarction induced by ischemia/reperfusion. Life Sci 80: 2076-2084.
- Abarbanell AM, Hartley JA, Herrmann JL, Weil BR, Wang Y, et al. (2011) Exogenous high-mobility group box 1 improves myocardial recovery after acute global ischemia/reperfusion injury. Surgery 149: 329-335.
- Zhang W, Han Y, Meng G, Bai W, Xie L, et al. (2014) Direct renin inhibition with aliskiren protects against myocardial ischemia/eeperfusion injury by activating nitric oxide synthase signaling in spontaneously hypertensive rats. J Am Heart Assoc 3: e000606.
- Yellon DM, Hausenloy DJ (2007) Myocardial reperfusion injury. N Engl J Med 357: 1121-1135.
- Koyanagi T, Noguchi K, Ootani A, Inagaki K, Robbins RC, et al. (2007) Pharmacological inhibition of epsilon PKC suppresses chronic inflammation in murine cardiac transplantation model. J Mol Cell Cardiol 43: 517-522.
- Mery PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R (1993) Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. J Biol Chem 268: 26286-26295.
- Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T (1993) Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl AcadSci U S A 90: 347-351.
- Shen W, Xu X, Ochoa M, Zhao G, Wolin MS, et al. (1994) Role of nitric oxide in the regulation of oxygen consumption in conscious dogs. Circ Res 75: 1086-1095.
- Shen W, Tian R, Saupe KW, Spindler M, Ingwall JS (2001) Endogenous nitric oxide enhances coupling between O2 consumption and ATP synthesis in guinea pig hearts. Am J Physiol Heart Circ Physiol 281: H838-H846.
- Bolli R (2001) Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning: an overview of a decade of research. J Mol Cell Cardiol 33: 1897-1918.
- Zaobornyj T, Valdez LB, Boveris A (2012) Effect of sildenafil on heart nitric oxide metabolism and mitochondrial function. In: Orrenius S, Packer L, Cadenas E (Eds) Mitochondrial signalling in health and disease. CRC Press, Taylor & Francis Group, USA, pp. 169-188.
- Deuse T, Koyanagi T, Erben RG, Hua X, Velden J, et al. (2010) Sustained inhibition of epsilon protein kinase C inhibits vascular restenosis after balloon injury and stenting. Circulation 122: S170-S178.
- Galli D, Carubbi C, Masselli E, Corradi D, Dei Cas A, et al. (2015) PKCε is a negative regulator of PVAT-derived vessel formation. Exp Cell Res 330: 277-286.