
Journal of Cardiobiology
Download PDF
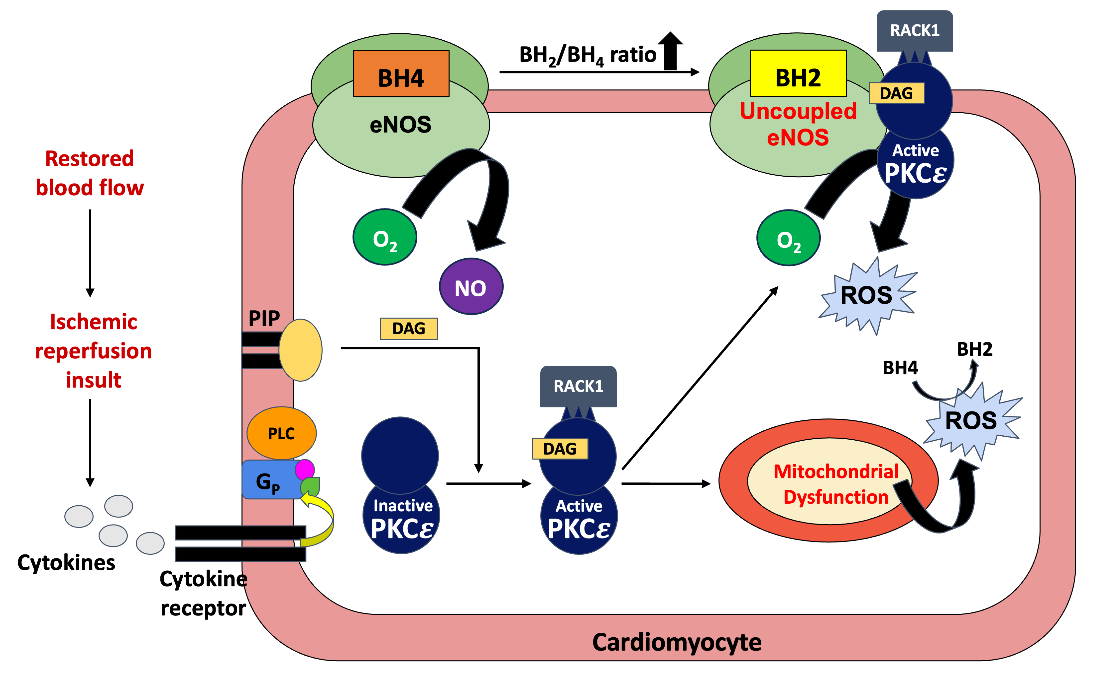 Figure 1:Mechanism of Protein Kinase C epsilon in MIR injury.
Figure 1:Mechanism of Protein Kinase C epsilon in MIR injury.
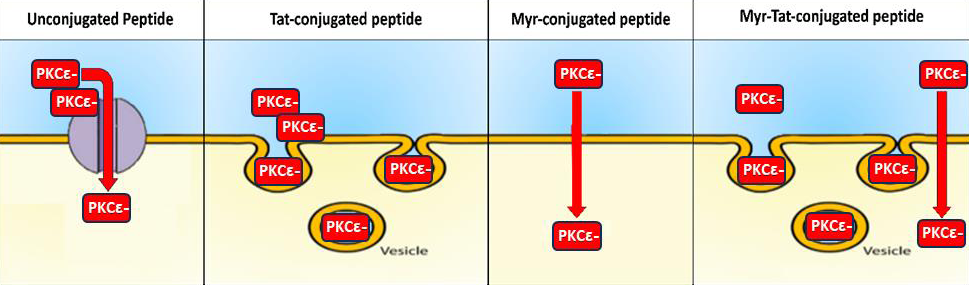 Figure 2:Proposed mechanisms of different drug conjugations in cell membrane penetration.
Figure 2:Proposed mechanisms of different drug conjugations in cell membrane penetration.
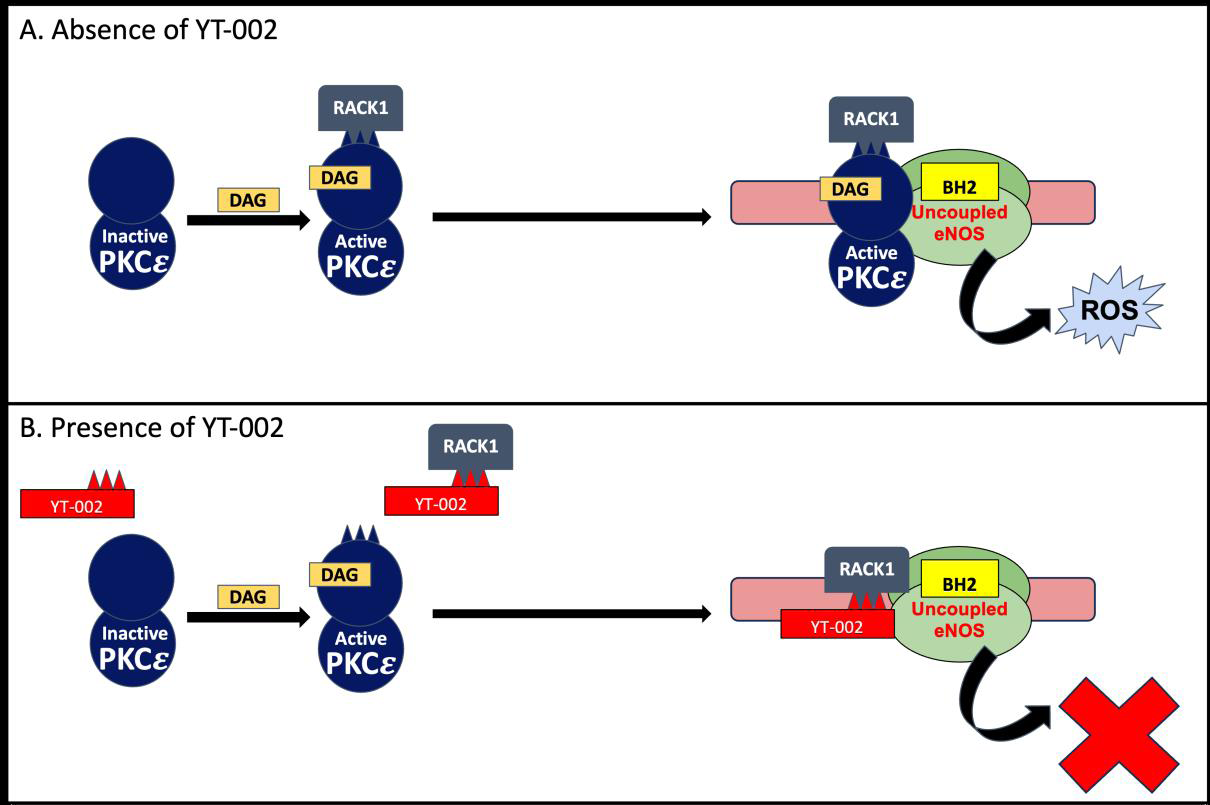 Figure 3:Mechanism of YT-002 in MIR injury. A. Normal pathophysiological response.
Figure 3:Mechanism of YT-002 in MIR injury. A. Normal pathophysiological response.
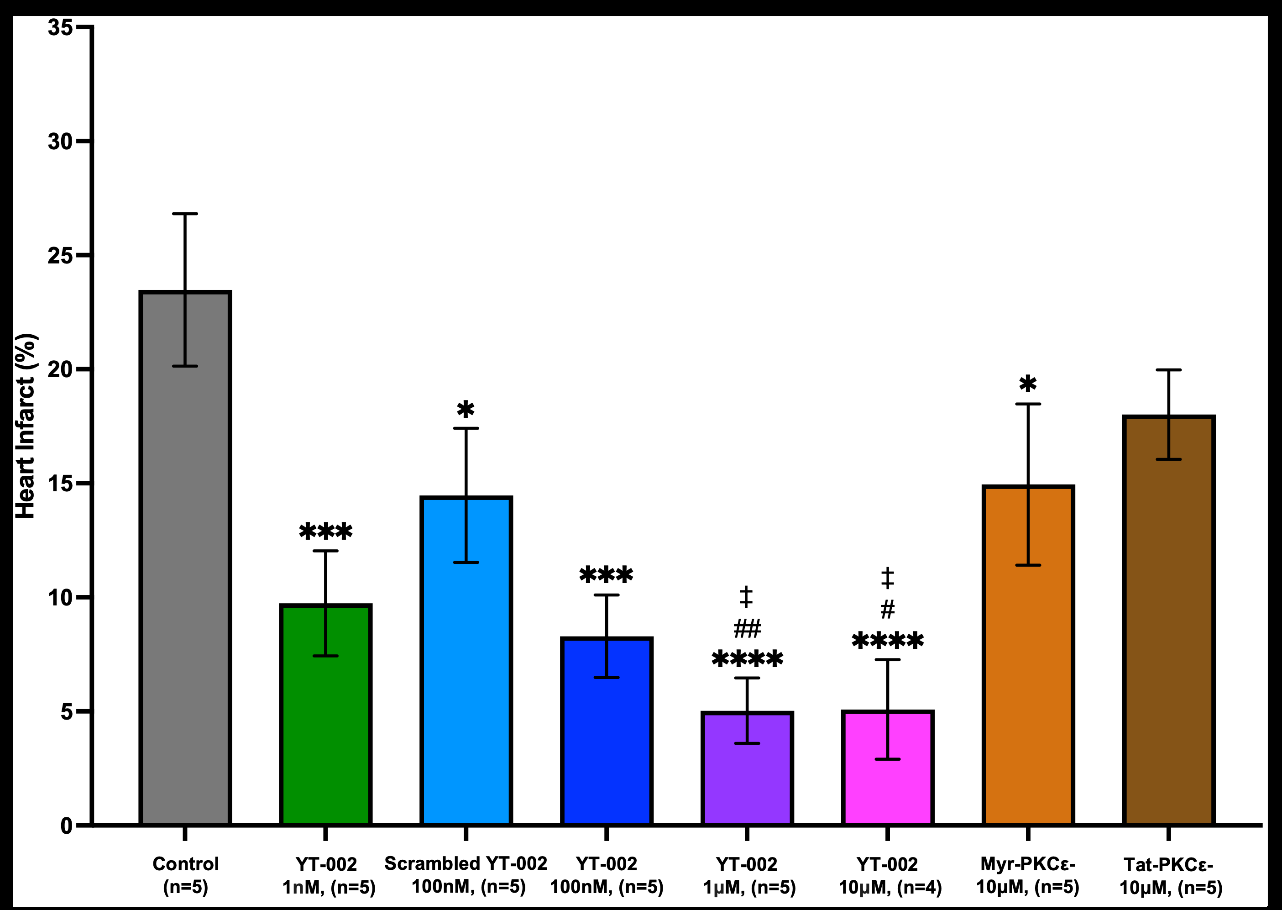 Figure 4:Heart infarct in an ex vivo rat heart MIR model.
Figure 4:Heart infarct in an ex vivo rat heart MIR model.
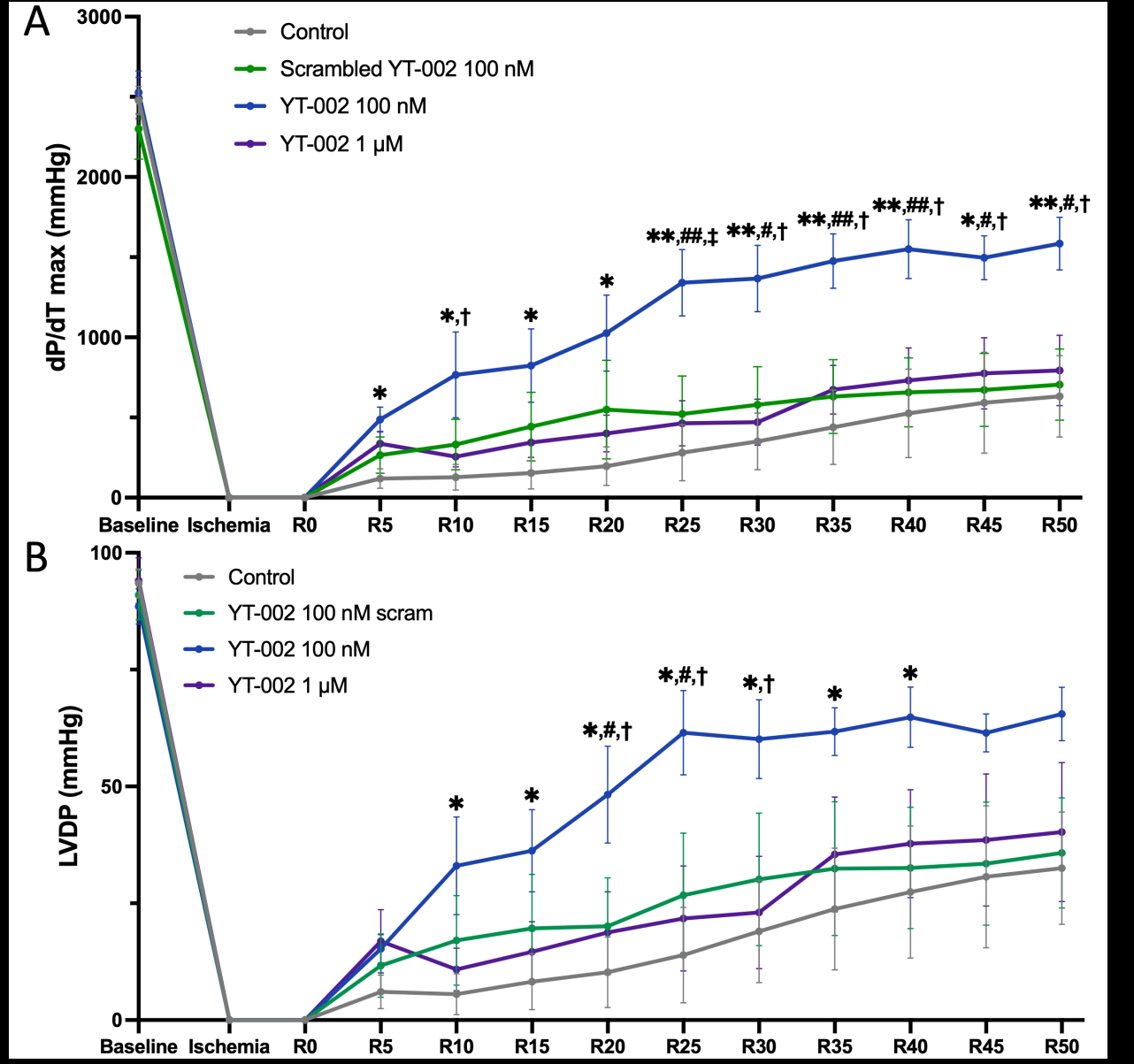 Figure 5: Cardiac function in an ex vivo rat heart MIR model. A. Time course of dP/dtmax values
Figure 5: Cardiac function in an ex vivo rat heart MIR model. A. Time course of dP/dtmax values
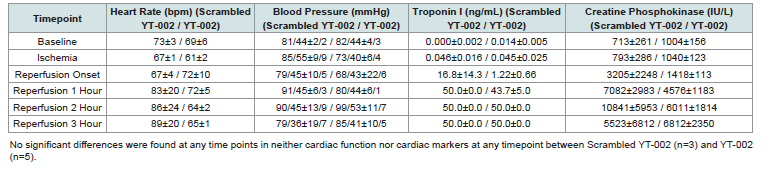 Table 1:Cardiac markers in an in vivo porcine heart MIR model.
Table 1:Cardiac markers in an in vivo porcine heart MIR model.
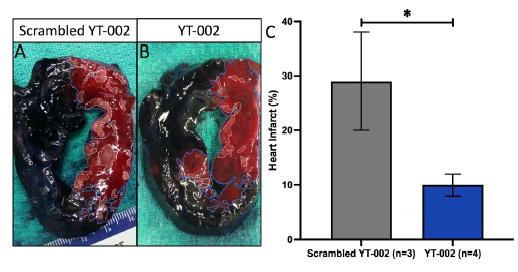 Figure 6:Heart infarct in an in vivo porcine heart MIR model. A, B Representative Heart Slices.
Figure 6:Heart infarct in an in vivo porcine heart MIR model. A, B Representative Heart Slices.
 Figure 7:Ejection Fraction for in vivo porcine heart MIR model.
Figure 7:Ejection Fraction for in vivo porcine heart MIR model.
Research Article
Cell Permeable Protein Kinase C Epsilon Peptide Inhibitor Mitigates Myocardial Ischemic- Reperfusion Injury
Nair A1, Tanoh DB1, Singh S2, Dean T2, Harrell K2, Melnik J2, Stinson C2, Le MA2, Humayun A2, Talukder Z2, Gazaway A3, Bryant A3, Chen Q2, Barsotti R2 and Young L1,2*
1Young Therapeutics, LLC, Philadelphia, PA, USA
2Philadelphia College of Osteopathic Medicine, Philadelphia, PA, USA
3Veranex, Atlanta, GA, USA
2Philadelphia College of Osteopathic Medicine, Philadelphia, PA, USA
3Veranex, Atlanta, GA, USA
*Address for Correspondence:Lindon Young, Young Therapeutics, LLC, 4170 City Avenue,
Philadelphia, PA, USA 19131; Telephone: 267 918 9373; Fax: 215 871 6869 Email Id: lindonyo@pcom.edu
Submission:26 November, 2024
Accepted:20 December, 2024
Published:27 December, 2024
Copyright: © 2024 Nair A, et al. This is an open access article distributed
under the Creative Commons Attribution License, which permits
unrestricted use, distribution, and reproduction in any medium, provided
the original work is properly cited.
Keywords:ATP-sensitive Mitochondrial Potassium Channels;
Uncoupled Endothelial Nitric Oxide Synthase; Reactive oxygen
species; Drug conjugation; Myocardial infarction (MI)
Abstract
Percutaneous coronary intervention is the primary treatment for
acute myocardial infarction (MI). Paradoxically, restoration of blood
flow causes myocardial ischemia reperfusion (MIR) injury, principally
due to the generation of reactive oxygen species (ROS). Protein Kinase
C epsilon (PKCε) is known to play a key role in ROS production. Our PKCε
inhibitor conjugated to myristic acid (Myr), YT-001, has shown efficacy
in reducing MIR injury in both ex vivo and in vivo animal models.
This study aims to evaluate the cardioprotective effects of a dualconjugated version of our PKCε inhibitor conjugated to Myr and a Transactivator of transcription (Tat), N-Myr-Tat-PKCε (YT-002), in ex vivo rat and in vivo porcine models of MIR injury.
Ex vivo rat hearts were subjected to 30 minutes of global ischemia followed by 50 minutes of reperfusion. In vivo porcine hearts underwent 1 hour of regional ischemia followed by 3 hours of reperfusion. YT-002, or a scrambled peptide control of YT-002, or a saline control was administered at reperfusion onset. Cardiac parameters were measured throughout reperfusion and infarct size was assessed postmortem.
In the ex vivo model, YT-002 (100nM) significantly decreased infarct size to 9.3±1.8%, (n=5, p<0.01) compared to saline control (23.4±3.3%, n=5) and significantly improved left ventricular function compared to saline and scrambled YT-002.
In the in vivo model, YT-002 (0.2mg/kg) significantly restored ejection fraction at the end of reperfusion (59.4±1.2%) to the baseline (59.4±0.8%, n=3, p=0.50) and reduced infarct size (10.0±2%, n=4) compared to scrambled YT-002 (29±9%, n=3; p<0.05).
These findings indicate that YT-002 can reduce cardiac infarct size and preserve cardiac function after MI. Since heart failure can correlate with infarct size, decrease MI-induced damage to the heart has the potential to decrease the severity of heart failure thus improving patient outcomes post-MI.
This study aims to evaluate the cardioprotective effects of a dualconjugated version of our PKCε inhibitor conjugated to Myr and a Transactivator of transcription (Tat), N-Myr-Tat-PKCε (YT-002), in ex vivo rat and in vivo porcine models of MIR injury.
Ex vivo rat hearts were subjected to 30 minutes of global ischemia followed by 50 minutes of reperfusion. In vivo porcine hearts underwent 1 hour of regional ischemia followed by 3 hours of reperfusion. YT-002, or a scrambled peptide control of YT-002, or a saline control was administered at reperfusion onset. Cardiac parameters were measured throughout reperfusion and infarct size was assessed postmortem.
In the ex vivo model, YT-002 (100nM) significantly decreased infarct size to 9.3±1.8%, (n=5, p<0.01) compared to saline control (23.4±3.3%, n=5) and significantly improved left ventricular function compared to saline and scrambled YT-002.
In the in vivo model, YT-002 (0.2mg/kg) significantly restored ejection fraction at the end of reperfusion (59.4±1.2%) to the baseline (59.4±0.8%, n=3, p=0.50) and reduced infarct size (10.0±2%, n=4) compared to scrambled YT-002 (29±9%, n=3; p<0.05).
These findings indicate that YT-002 can reduce cardiac infarct size and preserve cardiac function after MI. Since heart failure can correlate with infarct size, decrease MI-induced damage to the heart has the potential to decrease the severity of heart failure thus improving patient outcomes post-MI.
Abbreviations:
Area at risk (AR); Area of necrosis (AN); Cardiac output (CO);
Calcium (Ca2+); Diacylglycerol (DAG); Dihydrobiopterin (BH2);
Ejection Fraction (EF); Endothelial-derived nitric oxide (eNOS);
Intravenously (IV); Ischemia reperfusion (I/R); Left anterior
descending (LAD); Left ventricular developed pressure (LVDP); Left
ventricular end-diastolic pressure (LVEDP); Left ventricular enddiastolic
volume (LVEDV); Left ventricular end-systolic pressure
(LVESP); Left ventricular end-systolic volume (LVESV); Maximal
rate of decrease in left ventricular pressure (dP/dtmin); Maximal rate of
increase in left ventricular pressure (dP/dtmax)., Myocardial infarction
(MI); Myocardial Ischemia Reperfusion (MIR); Myristic acid (Myr)
Myr-PKCε inhibitor (YT-001); Myr-Tat-PKCε inhibitor (YT-002);
Nitric Oxide (NO); Protein Kinase C (PKC); Protein Kinase C Epsilon
(PKCε); Pulmonary capillary wedge pressure (PCWP); Reactive
Oxygen Species (ROS); Receptor for activated C kinase 1 (RACK1)
Superoxide (SO); Tetrahydrobiopterin (BH4); Transactivator of
transcription (Tat); Tumor Necrosis Factor Alpha (TNFa).
Introduction
Myocardial infarction (MI) is a major global health concern and
burden, with approximately 800,000 heart attacks occurring annually
in the United States [1-3]. MI is caused by partial or complete
blockage of a coronary artery, and it can be fatal or lead to long-term
health consequences. Arterial blockage prevents oxygenated blood
from reaching the distal regions of the myocardium (i.e., apex),
causing ischemia. Prolonged ischemia leads to dysfunction of ATP
production from mitochondria, leading to dysregulation of Na+/
K+ATPase that ultimately causes an increase in intracellular calcium
(Ca2+), which results in permanent muscle hypercontracture called
rigor[4-7]. This rigor condition compresses local blood vessels, further
limiting blood flow to the affected ischemic region, and it is principally
responsible for initial cardiac muscle infarction [8,9]. Reperfusion, is
the reintroduction of oxygenated blood to the myocardium and the
first priority of clinicians treating MI. While restoring blood flow
reduces the total amount of heart infarct, it paradoxically still causes
additional infarct damage [8,10]. This damage is known as myocardial
ischemia reperfusion (MIR) injury, and it is currently thought to be
responsible for approximately 50% of the infarct damage to the heart
[11,12]. Therefore, identifying therapeutic agents that can block/
inhibit reperfusion injury and mitigate damage from MIR is critical
to improving the outcomes after MI.
The mechanism of MIR injury has been well studied. The
restoration of oxygenated blood flow to the previously ischemic
myocardium triggers the release of tumor necrosis factor-alpha
(TNFα) within 15 minutes [13-15]. TNFα binds to its Gq-coupled
receptors and drive the production of the second messengers,
Ca2+and diacylglycerol (DAG), upon activation [13]. These second
messengers activate multiple different protein kinase C (PKC)
isoforms that phosphorylate a variety of protein targets. PKC epsilon
(PKCε) is especially sensitive to DAG and, when activated, promotes
mitochondrial ATP-sensitive K+ channel opening and uncoupled
endothelial nitric oxide synthase (eNOS) activity. It is well-recognized
that mitochondrial ATP-sensitive K+ channels and uncoupled eNOS
are two of the four primary sources of reactive oxygen species (ROS)
in the myocardium, making them key players in reperfusion injury
[Figure 1] [16-20]. ROS contributes to cellular damage through
mechanisms such as lipid peroxidation, protein modification, and
DNA damage, exacerbating inflammatory responses and promoting
apoptotic pathways [21-23]. Additionally, ROS impair endothelial
function, leading to increased vascular permeability and enhanced
leukocyte recruitment that amplifies inflammatory cascades associated
with reperfusion injury [8,24]. The opening of mitochondria ATPsensitive
K+ channels results in greater ROS production. These
ROS contribute to sustained mitochondrial permeability transition
pore opening, which can cause mitochondrial swelling, more ROS
production, and even cell death [8,24,25]. Normally, eNOS produces
nitrous oxide (NO) from oxygen and L-arginine using the necessary
cofactor tetrahydrobiopterin (BH4). During ischemic-reperfusion
(I/R) injury, ROS produced from the mitochondria and other sources
oxidize BH4 to dihydrobiopterin (BH2). The increased BH2/BH4 ratio
causes eNOS to enter an uncoupled state that interacts with PKCε
to instead produce superoxide (SO) [16,26]. Overall, PKCε’s role in
ROS generation makes it an ideal therapeutic target for attenuating
I/R injuries [27-29].
The restoration of blood flow after an ischemic period causes PKCε
activation through DAG production. RACK1, the receptor for active
C kinase specific to PKCε, translocates PKCε to the mitochondria
ATP-sensitive K+ channels and uncoupled eNOS, producing ROS.
In vitro studies of isolated mitochondria showed that PKCε only
stimulates ROS production in the presence of ATP-sensitive K+
channels [19,30]. This ROS production was then inhibited by a PKC
epsilon-specific inhibitor peptide epsilonV1-1(EAVSLKPT) [27-29,31]. Endothelial-derived NO plays an important role in creating anti-thrombotic surfaces, which reduces leukocyte-endothelial
interactions during inflammatory responses. Studies have shown that
ischemia causes a decrease in endothelial NO production, while cell
adhesion molecule expression and neutrophil infiltration are increased
during reperfusion [32]. eNOS uncoupling inhibits NO production
and promotes ROS generation. PKCε modulators affect NO and
SO levels through their interactions with eNOS. In rat mesenteric
postcapillary venules, BH2 significantly augmented leukocyte rolling,
adherence, and transmigration with or without a myristic acid
(Myr) conjugated PKCε activator (N-Myr-HDAPIGYD, Myr-PKCε
activator). A Myr-PKCε activator with BH4 and a Myr conjugated
PKCε inhibitor (N-Myr-EAVSLKPT, Myr-PKCε inhibitor, YT-
001), with or without BH2, both significantly reduced BH2-induced
inflammation [33,34]. YT-001 has been shown to significantly
reduce polymorphonuclear leukocyte (PMN)-induced postreperfused
cardiac contractile dysfunction as well as PMN adherence,
infiltration, and ICAM-1 expression in isolated rat hearts subjected
to I/R conditions [28]. The reduced NO production after ischemia
may be due to eNOS uncoupling. (N-Myr-HDAPIGYD, Myr-PKCε
activator). These results corroborated an earlier I/R study using
in vivo femoral veins [27]. They found that a Myr-PKCε activator with
BH2 significantly increased hydrogen peroxide production compared
to BH2 alone, and a Myr-PKCε activator with BH4 reversed this effect
[28]. They also found that YT-001 significantly increased NO release
in rat renal veins after extracorporeal shock wave lithotripsy [29].
Previous studies demonstrated that YT-001 significantly reduced
infarct size in ex vivo isolated perfused rat hearts and in an in vivo
porcine MIR model [35,36].
Cardiac transplantation is the most effective treatment for end-
stage heart failure. Although the one-year survival rate for cardiac
grafts after heart transplantation has improved, chronic rejection
remains a main cause of mortality[37]. Inhibiting PKC isozymes
improves cardiac graft survival after transplantation in animal
models, and conjugating PKC inhibitors with Tat, a thirteen amino
acid sequence (YGKKKRRQRRR), has shown improved efficacy. The
positively charged components of Tat interact with the negatively
charged components of the membrane, enabling peptide cargo
intracellular delivery via endocytosis, whereas Myr conjugation
facilitates intracellular delivery via simple diffusion [38-40]. A
Tat-conjugated PKCε inhibitor (Tat-PKCε inhibitor) has been
shown to extend graft survival and significantly improve functional
recovery in a cardiac transplantation model [37]. It also decreased
the inflammatory response by reducing T-cell and macrophage
infiltration and inhibiting mononuclear inflammatory cell adhesion
to the arterial wall. In models of cardiac transplantation and
angiotensin-induced heart failure, Tat-PKCε inhibitors preserved
the cardiac tissue architecture by reducing luminal narrowing and
preventing parenchymal fibrosis[37,41]. The ability of Tat-PKCε
inhibitor to improve cardiac graft and transplantation success, where
ROS reduce graft cell survival, makes it an appealing conjugation
to use in MIR models[37]. As a result, a combination of Tat and
Myr conjugation may result in an overall increase in drug delivery
compared to either conjugation alone [Figure 2].
Unconjugated peptides must enter cells via the facilitated
diffusion, requiring a carrier protein. Tat conjugation enables
intracellular delivery through endocytosis, and Myr conjugation
enables simple diffusion. Myr-Tat conjugation aims to increase
intracellular delivery by combining these cargo delivery mechanisms.
(adapted [40]).
We have developed a PKCε inhibitor conjugated to both Myr and
Tat (N-Myr-Tat-CC-EAVSLKPT), named YT-002, to test whether
this dual conjugation will improve PKCε inhibition. YT-002 binds
to the scaffolding protein Receptor for activated C kinase1 (RACK1),
which normally transports PKCε to mitochondrial ATP-sensitive
K+ channels and eNOS [31]. As previously mentioned, these two
substrates produce ROS during reperfusion, so YT-002’s activity
prevents ROS production [Figure 3]. Previous therapeutics targeting
I/R injury reduction failed primarily because they aim to scavenge
already formed damaging free radicals.YT-002 instead works to
prevent the further generation of new ROS.
Active PKCε is translocated by RACK1 to uncoupled eNOS,
leading to excessive ROS production. These ROS contribute to heart
infarct and cardiac dysfunction seen in MI. B. Pharmacological
mechanism of YT-002. YT-002 binds to RACK1, preventing PKCε
translocation. This inhibition reduces ROS production and mitigates
its negative downstream effects. (adapted from [31]).
We hypothesize that YT-002 will show increased potency
compared to YT-001 or Tat-PKCε inhibitor in our ex vivo rat heart
MIR model. We also predict that YT-002 will significantly reduce
infarct size and improve cardiac parameters compared to saline
control. When tested in an in vivo pig MIR model, we expect that YT-
002 will significantly reduce infarct size and improve cardiac function
compared to its scrambled version.
Methods
All experimental protocols were approved by the Institutional
Animal Care and Use Committee and performed in accordance with
the institutional policies of the Philadelphia College of Osteopathic
Medicine and the Government Center for Medical Intervention
(Veranex, formally T3 Labs)(Atlanta, GA) an Association for
Assessment and Accreditation of Laboratory Animal Care-approved
facility.
Drug Conjugation:
A PKCε inhibitor peptide (EAVSLKPT) was conjugated to both
Myr and Tat to create a dual-conjugated PKCε inhibitor peptide
(N-Myr-Tat-CC-EAVSLKPT) we have named YT-002. A disulfide
bond (CC) was placed between the conjugations and peptide
sequence to improve RACK binding. Single-conjugated versions of
this drug (YT-001 and N-Tat+EAVSLKPT) were also created and
tested. A scrambled version of the inhibitor peptide (LSETKPAV)
was also conjugated to Myr and Tat to create the scrambled version
of YT-002 (N-Myr+Tat+LSETKPAV) to serve as a control peptide
(Genemed Synthesis, Inc, San Antonio TX 78249). All drugs were
diluted to the desired concentration between 100pM and 10μM using
0.03% DMSO in the perfusate.
Ex-Vivo Rat MIR Model:
Male Sprague-Dawley rats (275-325 g, Charles River, Springfield,
MA) were housed in a 12-hour light and 12-hour dark cycle in a
temperature-controlled room. They were given continuous access to
food and water. All work with these rats followed our animal-handling
protocol A21-002. Rats were anesthetized via an intraperitoneal
injection of sodium pentobarbital (60mg/kg) and heparin (1000
units). Hearts were excised and placed on a Langendorff perfusion
machine [36] where they were perfused with Krebs’ buffer at 37oC at
80 mmHg constant pressure. After a 15-minute stabilization period
at which baseline measurements were made, hearts were subjected to
30 minutes of global ischemia followed by 50 minutes of reperfusion.
Heart rate, left ventricular end-systolic pressure (LVESP), left
ventricular end-diastolic pressure (LVEDP), the maximal rate of
increase in left ventricular pressure (dP/dtmax), and the maximal rate
of decrease in left ventricular pressure (dP/dtmin) were measured using
an SPR-524 pressure catheter (Millar Instruments, Inc., Houston, TX)
placed in the left ventricle. Data values were acquired and stored using
a PowerLab/8Sp data acquisition system (AD Instruments, Colorado
Springs, CO). Left ventricular developed pressure (LVDP) was found
by subtracting the LVEDP from the LVESP (LVESP-LVEDP). The
chosen drug or saline control was delivered during the first 5 minutes
of reperfusion using a syringe pump at a rate of 1 mL/min.
After the reperfusion period, the hearts were removed from the
Langendorff machine and placed in a -20°C freezer for 30 minutes.
The hearts were then sectioned perpendicular to their long axis
into seven 2-millimeter slices. These slices were incubated in 1%
2,3,5-triphenyltetrazolium chloride (TTC) in 0.2 M Tris buffer
(pH=7.41) for five minutes to delineate infarct (pale color) and viable
heart tissue (red color). They were placed in 4% paraformaldehyde
solution to improve contrast and then photographed. The slices were
dissected to separate the infarct and viable tissue, and the percent of
infarct was found by dividing the weight of the infarct tissue by the
total weight of the tissue.
All data are presented as mean±SEM. ANOVA analysis using
Fisher’s PSLD test was used to assess any statistical differences in
infarct and cardiac parameters between groups. Probability values
less than 0.05 were considered statistically significant.
In vivo porcine MIR model:
Castrated Male Yorkshire pigs (35-50kg) were purchased by T3
labs. All work with these pigs followed our animal-handling protocol
YF01P.Pigs were housed for at least 72 hours before surgery to allow
acclimation to the environment, and each animal underwent a general
visual health survey by a T3 Labs veterinarian to ensure proper health
before surgery. Aspirin (anticoagulant, 300mg) and Amiodarone
(antiarrhythmic, 800mg) were given once a day orally for 1 and 3
days, respectively, before surgery. Ketamine (15mg/kg) and xylazine
(1mg/kg) were administered through intramuscular injection to
sedate the pig. Propofol was given intravenously (IV) for laryngeal
relaxation (2-4mg/kg) as needed. The pig was then intubated and
maintained using Isoflurane (1.5-2.5%, inhalant) and either lactated
ringer solution or 0.9% NaCl (2.5-5 mL/kg/hr, IV).
Pigs were subjected to 1 hour of regional ischemia by ballooncatheter
inflation, which was then deflated for 3 hours of reperfusion
A catheter with fluoroscopic guidance was used to place the balloon
catheter at the level of the second diagonal branch of the left anterior
descending (LAD) coronary artery, or approximately 40% of the left
ventricular anterior wall distance from the apex to the base of the
heart, to cause an antero-apical MI. Ten minutes before ischemia,
antiarrhythmic agents’ amiodarone (5mg/kg) and lidocaine (2mg/
kg) were administered intravenously. End-tidal CO2 levels, pulse
oximetry, ECG, pulmonary capillary wedge pressure (PCWP), rectal
temperature, heart rate, blood pressure, and depth of anesthesia were
measured during ischemia. The balloon catheter was also used for
drug administration and blood pressure measurement. Catheterballoon
deflation marked the onset of reperfusion, and either YT-002
or Scrambled YT-002 was administered into the LAD. A 0.2mg/kg
concentration was used to approximate the 1μM concentration of
YT-002 tested in our ex vivo rat heart model.
Additional catheters were placed in the pulmonary artery and
femoral artery through external jugular vein sheaths to measure
PCWP and flow, respectively. Two-dimensional electrocardiography
was used to measure left ventricular end-diastolic volume (LVEDV)
and left ventricular end-systolic volume (LVESV) using a modified
Simpson’s rule technique. Blood samples were taken from the arterial
sheath to measure Troponin I and Creatine Phosphokinaselevels,
and they were analyzed by Antech Diagnostics (Atlanta, GA).
Electrocardiograph measurements, blood pressure measurements,
and blood samples were taken at baseline (15 min before ischemia),
30 minutes into ischemia, and every hour during reperfusion.At
the end of the reperfusion period, the chest cavity was opened, and
the coronary artery, aorta, pulmonary artery, and caudal vena were
occluded. We ensured to occlude the coronary artery at the same
place where the balloon-catheter was placed.
The left atrium was incised, and 1% Evans Blue Dye (1 ml/kg) was
injected into the left atrium to determine the area that was not at risk
by labeling the area not at risk in blue. The pig was then euthanized
using Potassium Chloride (1-2 mEq/kg) via IV injection, and the
heart was excised. The left ventricle was cut axially into 8-millimeter
slices and then placed in 1% TTC for 1 hour to determine the area
at risk (AR). Thereafter, the area of necrosis (AN) was the area that
turned pale after the TTC staining. The slices were photographed,
and three blinded analysts measured the AR and AN. A percent
infarct was calculated using AN/AR, and the analyst’s values were
averaged to obtain a final percent infarct for each heart using ImageJ
analysis. Ejection fraction (EF) was calculated by dividing stroke
volume (difference of end-systolic volume and end-diastolic volume)
by end-diastolic volume and cardiac output (CO) was calculated by
multiplying the heart rate by the stroke volume.
All data are presented as mean±SEM. Student’s t-test was used
to assess any statistical differences in infarct and cardiac parameters
between groups. Probability values less than 0.05 were considered
statistically significant
Results
Ex-vivo results:
YT-002 was tested at concentrations ranging from100pM to
10μM. YT-002 significantly reduced infarct size compared tosaline
control (23.5±1.8%, n=5) at concentrations of 1nM (9.7±2.3%,
n=5, p=0.006), 100nM (9.3±1.8%, n=5, p=0.0002), 1μM. (5.0±2.0%,
n=5, p<0.001), and 10μM (5.1±2.2%, n=5, p<0.0001). Interestingly,
Scrambled YT-002 (100nM) also significantly decreased infarct
(14.5±2.9%, n=5, p=0.0175) compared to control. Myr-PKCε
inhibitor (10μM) [Figure 4]. (14.9±2.4%, n=5, p=0.0237) significantly
reduced infarct compared to control, whereas Tat-PKCε inhibitor
(10μM) (17.6±2.3%, n=5) did not have the same effect [Figure 4].
Comparing the PKCε inhibitor conjugations, YT-002 (1μM and
10μM) significantly reduced infarct compared to Myr-PKCε inhibitor
(10μM) (p=0.0095 and p=0.0144, respectively) and Tat-PKCε
inhibitor (10μM) (p=0.0018 and p=0.0030, respectively).
YT-002 1nM (9.7±2.3%, n=5), YT-002 100nM (8.3±1.8%, n=5),
Scrambled YT-002 100nM (14.5±2.9%, n=5), YT-002 1μM (5.0±1.4%,
n=5), and YT-002 10μM (5.1±2.2%, n=4) significantly reduced heart
infarct compared to saline control (23.5±3.3%, n=5). Myr-PKCε
inhibitor (Myr-PKCε-)10μM also significantly decreased heart infarct
(14.9±2.4%, n=5) compared to control while Tat-PKCε inhibitor
(Tat-PKCε-) 10μM (17.6±2.3%, n=5) did not. YT-002 1μM and
YT-002 10μM significantly decreased infarct size compared to these
singly conjugated PKCε inhibitors. * vs. control, # vs. Myr-PKCε-, †
vs. Tat-PKCε-. *p=0.0237 for Myr-PKCε-, *p=0.0175 for Scrambled
YT-002 100nM, ***p=0.0006 for YT-002 1nM, ***p=0.0002 for YT-
002 100nM, ***p<0.001 for YT-002 1μM, ***p<0.0001 for YT-002
10μM, #p=0.0144 for YT-002 10μM, ##p=0.0095 for YT-002 1μM, ‡p
=0.0030 for YT-002 10μM ‡p=0.0018 for YT-002 1μM
Two key measurements of left ventricular performance (i.e., the
heart’s ability to pump oxygenated blood) are dP/dtmax [Figure 5A]
and LVDP [Figure 5B]. YT-002 (100nM) significantly increased
dP/dtmax (p<0.05) from 5 minutes of reperfusion until the end of
reperfusion compared to saline control. YT-002 (100nM) also
significantly increased dP/dtmax (p<0.05) compared to Scrambled YT-
002 (100nM) and YT-002 (1μM) from the 25 minutes of reperfusion
until the end of reperfusion. YT-002 (100nM) significantly increased
LVDP (p<0.05) compared to saline control from 10 to 40 minutes
of reperfusion. YT-002 (100nM) also briefly significantly increased
LVDP (p<0.05) compared to Scrambled YT-002 (100nM) and YT-
002 (1μM) in the middle of reperfusion.
YT-002 (100nM) significantly increased dP/dtmaxvalues compared
to saline control throughout the reperfusion period. In addition, YT-
002 (100nM) significantly increased dP/dtmax compared to scrambled
(scram) YT-002 (100nM) and YT-002 (1μM) after 25 minutes of
reperfusion. B. Time course of LVDP values. YT-002 (100nM)
significantly increased LVDP compared to saline control from 10
to 40 minutes. It also significantly increased LVDP compared to
scram YT-002 (100nM) and YT-002 (1μM) during the mid-point of
the reperfusion period. * p<0.05, **p<0.01 for YT-002 (100nM) vs.
Control; #p<0.05, ##p<0.01 for YT-002 (100nM) vs. scram YT-002
(100nM); †p<0.05, ‡p<0.01 for YT-002 (100nM) vs. YT-002 (1μM).
“R#” on the x-axis indicates the reperfusion time in minutes.
In-vivo results:
Based on our rat heart experiments, we chose to use YT-002 1μM
(i.e. 0.2mg/kg) for testing in the porcine MIR model since this was
the lowest concentration that had tissue-salvaging effects without
impacting cardiac function, in contrast to YT-002 10μM which
depressed cardiac function relative to control (data not shown).
Scrambled YT-002 (0.2mg/kg) was used as our control. In our in
vivo pig heart experiments, YT-002 significantly reduced infarct
(10.0±2.0%, p=0.039) compared to Scrambled YT-002 (29.0±9.0%.)
[Figure 6]. YT-002 significantly increased EF at the second hour
(55.3±0.8% vs. 59.4±1.1%, p=0.02) and third hour (54.6±1.3% vs.
58.4±0.8%, p=0.04) of reperfusion compared to Scrambled YT-002
despite having similar reperfusion onset EF values [Figure 7]. YT-
002 also restored EF to its baseline value by the end of reperfusion
(59.4±1.2% vs. 59.4±0.8%, p=0.50) while Scrambled YT-002 failed to
have the same effect (62.0±0.58% vs. 55.3±0.9%, p=0.003). Heart rate,
blood pressure, creatine phosphokinase, and Troponin I were not
significantly different at any timepoint throughout this experiment
[Table 1].
Scrambled YT-002 (A) shows greater infarct compared to YT-
002(B). The area at risk (AR) is outlined in blue and the area of
necrosis (AN) is outlined in white. C. Graph of Heart Infarct. YT-
002 significantly reduced heart infarct (10.0±2.0%, n=4) compared
to Scrambled YT-002 (29.0±9.0%,n=3). Heart infarct = AN/AR
*p=0.039.
YT-002 (n=5) and Scrambled YT-002 (n=3) groups had similar
ejection fractions at baseline and the onset of reperfusion. YT-002
significantly increased ejection fraction compared to Scrambled YT-
002 at the second hour (55.3±0.8% vs. 59.4±1.1%, *p=0.02) and third
hour (54.6±1.3% vs. 58.4±0.8%, *p=0.04) of reperfusion. By the end
of reperfusion, YT-002 restored ejection fraction back to baseline
(59.4±1.2% vs. 59.4±0.8%, p=0.50) while Scrambled YT-002 did not
(62.0±0.58% vs. 55.3±0.9%, **p=0.003)
Discussion
Summary of Major Findings:
Our study aimed to determine if YT-002 would mitigate MIR
injury by reducing infarct size and improving cardiac function.
Additionally, we explored whether the dual conjugation of YT-002
(Myr+Tat) would be superior in reducing MIR injury in comparison
to Myr or Tat conjugation alone. We found that:
1. YT-002 demonstrated 10,000 times greater potency compared
to either YT-001 or Tat-PKCε inhibitor. In the ex vivo rat heart
model, YT-002 significantly reduced infarct size at 1nM, while YT-
001only caused a similar effect at 10μM and Tat-PKCε inhibitor had
no effect at 10μM.
2. YT-002 treatment significantly reduced infarct size compared
to its control in both ex vivo and in vivo MIR models.
3. YT-002 treatment significantly improved cardiac function
during reperfusion, as shown by improvements in dP/dtmax and
LVDP in the ex vivo rat heart model and EF in the in vivo pig model.
4. YT-002’s cardioprotective effects are amino-acid specific, as
its scrambled control failed to reduce infarct and restore cardiac
function as YT-002 did in the in vivo MIR pig model.
Peptide Conjugation:
The dual conjugation of YT-002 appears to be more effective at
penetrating cardiomyocytes and assisting the PKCε inhibitor than
its single conjugation versions. A Tat conjugated PKCε inhibitor
tested in Phase II clinical trials for postherpetic neuropathy and
postoperative orthopedic pain showed no safety or immunogenicity
concerns [38,39]. No Myr acid conjugated PKCε inhibitor has been
tested in clinical trials yet. However, Myr is an endogenous cargo
delivery molecule, suggesting it would present no safety concerns in a
clinical trial[40]. As a result, we anticipate that YT-002 is unlikely to
present any safety or immunogenic problems in clinical trials.
Mechanism of YT-002:
YT-002 operates through a unique mechanism of PKCε
inhibition by directly targeting the cellular mechanisms that
cause oxidative stress and subsequent myocardial damage during
reperfusion. It prevents the formation of ROS by stopping PKCε from
interacting with eNOS and mitochondria ATP-sensitive K+ channels
[17,19,27,28]. Unlike conventional treatments that neutralize ROS
after they are generated, YT-002 quenches the production of ROS
[8,27-29]. By specifically targeting PKCε, whose main activity
during reperfusion is in oxidative stress pathways, YT-002 provides
a focused approach that preserves myocardial tissue integrity and
function and minimizes off-target interactions,reducing the potential
for adverse effects compared to abroader-acting pharmacological
agent [27–29,33,35–37,41]. At the same time, YT-002 provides a dual
mechanism of action by inhibiting two ROS-generating pathways
Comparison of YT-002 to other PKCε Inhibitors:
Other PKCε inhibitors have already been tested in MIR animal
models. A novel Tat conjugated PKCε inhibitor was found to
significantly reduce infarct size and the number of ventricular
fibrillation cases in a murine cardiac transplantation model [37,41].
Our lab previously showed that the Myr conjugated PKCε inhibitor
(YT-001) significantly reduced infarct size and recovered dP/dtmax
values in an ex vivo rat heart model, and the same drug was found
to reduce infarct size and restore EF to baseline values in an in vivo
pig heart model [35,36]. While our experiments with YT-002 in these
same MIR models produced similar results, YT-001 had to be used at
concentrations between 10μM and 20μM, more than 10,000 times the
concentration of YT-002 found to be effective in our study. (Figure
4) [1nM]). These studies provide additional support that targeting
PKCεis an effective way to reduce MIR injury and that YT-002’s dual
conjugation enables it to be effective at lower concentrations.
Potential Role YT-002 in other I/R settings:
Our results demonstrate that YT-002 can mitigate I/R injury by
inhibiting ROS generation in the setting of MI. PKCε has been shown
to be present and even acutely increased during the I/R conditions
that occur during cardiac and kidney transplantation, cerebral stroke,
and extracorporeal shockwave lithotripsy, and all of these conditions
involve ROS damage [29]. Our YT-001 was shown to be effective
in reducing kidney I/R injury in vivo [42]. Given that the YT-002’s
mechanism of action is independent of the tissue setting, this drug
has the potential to be effective in reducing I/R injury in a variety of
ischemic organ settings.
Study Limitations:
This study presents limitations in its attempt to provide preclinical
knowledge. First, our Troponin I measurement tool did
not have an adequately large range for our experiment, resulting
in all measurements at or above 50 ng/mL being recorded as 50
ng/mL. Troponin I is widely used in the clinic for assessing MI, so
this limitation prevented us from further understanding YT-002’s
potential in mitigating MIR injury. Pigs and rats are known to have
different cardiac properties than humans, though pigs anatomically
and physiologically are more similar to humans [43]. These
differences provide limitations in our ability to use these animal
MIR models to predict a drug’s success in the clinic. Both models
caused MI through immediate methods whereas MI has a gradual
pathogenesisin humans [43,44]. In addition, these animal models
only used PKCε inhibition to treat the induced MI, whereas our
drug would likely be an adjunctive therapy to treatments currently
used in the clinic, such as percutaneous coronary intervention. A
MIR model that more closely resembles human MI in theseways will
likely produce more translatable results.Both experiments also only
used male animals, making our findings less applicable to the general
population. In addition, MI has been shown to have long-term effects
on patients, while all our analyses were done during or immediately
after reperfusion.
Future Studies:
We aim to conduct a six-month in vivo porcine MIR survival
study to evaluate the capabilities of YT-002 to mitigate the long-term
effects of MI. This study will also help determine the therapeutic
window and dosing regimen that optimizes efficacy and minimizes
potential adverse effects in a heart model that closely mimics human
cardiac physiology. We will use the same MIR pig model presented
in this manuscript, andleft ventricle EF and high-sensitivity troponin
and creatine phosphokinase-MB will be measured throughout the
six-month survival periodto determine if YT-002 can reduce post-MI
incident heart failure.
Conclusion
In conclusion, YT-002 was cardioprotective and reduced MIR
injury in ex vivo and in vivo animal MIR models. It significantly
reduced infarct size and restored LV function. This effect resulted from
YT-002 inhibiting PKCε-driven ROS generation at the beginning of
reperfusion, suggesting it could be effective in other I/R scenarios.
Our results principally highlight YT-002’s potential for improving
MI outcomes in the clinic. Future studies will evaluate the long-term
effects of YT-002 treatment in a porcine MIR model, and we aim to
enter this drug into Phase 1 clinical trials.
Highlights:
-Myocardial ischemia reperfusion injury is a major contributor
to myocardial infarction pathophysiology.
- YT-002 mitigates this injury by inhibiting the production of
reactive oxygen species.
- YT-002 administration at reperfusion onset reduces infarct
size and restores LV function in MIR.
- Phase 1 clinical trials are next in studying YT-002’s potential
as an MI therapeutic.
Acknowledgements
Grants: This work was funded by a grant from the National Heart,
Lung, and Blood Institute. Grant#: 1R43HL160338-01
We would like to acknowledge Uzma Gullabzada, Mansoor
Gullabzada, and Bansari Patel (Insight Hospital, Chicago, IL) for
organization help with this manuscript. Dr. Thomas Argentieri,
Young Therapeutics, LLC, BD & Scientific Advisor with editing the
manuscript.
Arjun Nair performed data analysis and interpretation, wrote
and edited the manuscript, and approved the final version to be
published. Desmond Boakye Tanoh collected data, performed data
analysis and interpretation, wrote and edited the manuscript, and
approved the final version to be published. Sunit Singh performed
data analysis and interpretation. Tameka Dean assisted with data
interpretation and revised our work for important intellectual
content. Kayla Harrell, Juliet Melnik, Cameron Stinson, and Annam
Humayun performed data analysis and interpretation, and Juliet
Melnik also approved the final version to be published. Mai An
Le and Zinya Talukder assisted with manuscript writing. Ashley
Gazaway and Antwawn Bryant assisted with experimental design and
data collection. Qian Chen and Robert Barsotti revised our work for
important intellectual content and approved the final version to be
published. Lindon Young led the experimental design, collected data,
performed data interpretation, wrote and edited the manuscript,
revised our work for important intellectual content, and approved
the final version to be published. All authors agree to be accountable
for all aspects of the work in ensuring that questions related to
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
The authors of this manuscript have no relationship with industry.
No disclosure is required. Involved Institutions: Philadelphia College
of Osteopathic Medicine, 4170 City Ave, Philadelphia, PA 19131;
Veranex, Atlanta, GA. Government Center for Medical Intervention
References
Citation
Nair A, Tanoh DB, Singh S, Dean T, Harrell K, et al. Cell Permeable Protein Kinase C Epsilon Peptide Inhibitor Mitigates Myocardial Ischemic-Reperfusion Injury. J Cardiobiol. 2024;8(1): 1.