
Journal of Clinical and Investigative Dermatology
Download PDF
Research Article
*Address for Correspondence: William W. Huang, Center of Dermatology Research, Department of Dermatology ; Wake Forest University School of Medicine; Winston- Salem, North Carolina, Tel: (336) 716-2198; Fax: (336) 716-7732; E-mail: whuang@wakehealth.edu
Citation: Shokeen D, O’Neill JL, Davis SA, Moustafa F, Huang WW. Characterizing the Treatment of Autoimmune Bullous Disorders from 1993 through 2010: a NAMCS Study. J Clin Investigat Dermatol. 2013;1(1): 4.
Copyright © 2013 Huang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Investigative Dermatology | ISSN 2373-1044 | Volume: 1, Issue: 1
Submission: 25 October 2013 | Accepted: 28 November 2013 | Published: 30 November 2013
Reviewed & Approved by: Dr. Martin Steinhoff Professor of Dermatology & Surgery, Deptartment of Dermatology at UCSF.

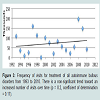
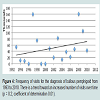
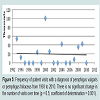
 There was a trend toward an increased number of outpatient visits for treatment of all autoimmune bullous diseases from 1993 to 2010 (Figure 3). Visits to physician offices for bullous pemphigoid (Figure 4) or for pemphigus (Figure 5) from 1993 to 2010 also did not increase significantly.
There was a trend toward an increased number of outpatient visits for treatment of all autoimmune bullous diseases from 1993 to 2010 (Figure 3). Visits to physician offices for bullous pemphigoid (Figure 4) or for pemphigus (Figure 5) from 1993 to 2010 also did not increase significantly.
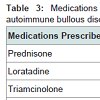
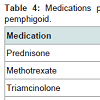
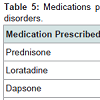
 Prednisone, dapsone, and dexamethasone were the most frequently prescribed medications by all physicians for the treatment of all autoimmune bullous disorders (Table 2). For all patients diagnosed with an autoimmune bullous disease, dermatologists prescribed prednisone, loratadine, and triamcinolone most frequently (Table 3). For bullous pemphigoid, the most frequently prescribed medications were prednisone, methotrexate, and triamcinolone, prescribed to 42, 13 and 11% of patients, respectively (Table 4).Patients with pemphigus vulgaris or foliaceus were treated most frequently with prednisone, dapsone and loratadine, prescribed to 49, 15, and 10% of patients, respectively (Table 5).
Prednisone, dapsone, and dexamethasone were the most frequently prescribed medications by all physicians for the treatment of all autoimmune bullous disorders (Table 2). For all patients diagnosed with an autoimmune bullous disease, dermatologists prescribed prednisone, loratadine, and triamcinolone most frequently (Table 3). For bullous pemphigoid, the most frequently prescribed medications were prednisone, methotrexate, and triamcinolone, prescribed to 42, 13 and 11% of patients, respectively (Table 4).Patients with pemphigus vulgaris or foliaceus were treated most frequently with prednisone, dapsone and loratadine, prescribed to 49, 15, and 10% of patients, respectively (Table 5).
Characterizing the Treatment of Autoimmune Bullous Disorders from 1993 through 2010: a NAMCS Study
Divya Shokeen, Jenna L. O’Neill, Scott A. Davis, Farah Moustafa and William W. Huang*
- Center of Dermatology Research, Department of Dermatology ; Wake Forest University School of Medicine, USA
*Address for Correspondence: William W. Huang, Center of Dermatology Research, Department of Dermatology ; Wake Forest University School of Medicine; Winston- Salem, North Carolina, Tel: (336) 716-2198; Fax: (336) 716-7732; E-mail: whuang@wakehealth.edu
Citation: Shokeen D, O’Neill JL, Davis SA, Moustafa F, Huang WW. Characterizing the Treatment of Autoimmune Bullous Disorders from 1993 through 2010: a NAMCS Study. J Clin Investigat Dermatol. 2013;1(1): 4.
Copyright © 2013 Huang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Investigative Dermatology | ISSN 2373-1044 | Volume: 1, Issue: 1
Submission: 25 October 2013 | Accepted: 28 November 2013 | Published: 30 November 2013
Reviewed & Approved by: Dr. Martin Steinhoff Professor of Dermatology & Surgery, Deptartment of Dermatology at UCSF.
Abstract
Autoimmune blistering diseases, though uncommon, are associated with considerable morbidity. There is no established therapeutic ladder for the treatment of blistering disorders. Data from the National Ambulatory Medical Care survey from 1993 to 2010 was analyzed to determine the number of visits for blistering diseases, patient demographics, the specialty of treating physicians, and the most frequently prescribed treatments. Patients with autoimmune blistering disorders were primarily elderly, female, and non-Hispanic white. The majority of patients were managed by dermatologists. There has been no significant change in the number of patients diagnosed with bullous diseases over the observed time period. Oral prednisone was the primary treatment prescribed for patients with both bullous pemphigoid and pemphigus. Though prednisone is the primary medication prescribed for patients with blistering disorders, immunosuppressive therapies are associated with increased morbidity in elderly patients. Alternative therapies should be considered for the treatment of autoimmune bullous disorders when feasible taking into account the associated risks and side effect profile of these medications.Keywords
Bullous pemphigoid; Pemphigus vulgaris; Dermatology; Treatment; AutoimmuneIntroduction
Autoimmune blistering disorders, which include bullous pemphigoid and disorders within the pemphigus family, are associated with considerable morbidity and impact on quality of life. Bullous lesions on skin and mucous membranes are induced by autoantibodies targeting components of the epidermis or Dermo- Epidermal Junction (DEJ). Afflicted patients may also have severe manifestations in other organ systems resulting in oral and laryngeal ulcers, esophageal strictures, and ocular scarring resulting in blindness [1]. The clinical diagnostic criteria for blistering diseases are not well characterized and accurate diagnosis often relies upon histopathologic and immunohistochemical evaluation of tissue specimens.Treatment of blistering diseases aims to suppress the autoimmune response and may include topical or oral medications [2]. The mainstay of treatment is oral corticosteroids and additional immunosuppressive agents are often added to avoid the numerous potential adverse effects associated with long term oral corticosteroid treatment. Commonly employed immunosuppressive agents include methotrexate, dapsone, azathioprine, and mycophenolate mofetil. Treatment with these agents may be associated with morbidity independent of the disease state, and long term therapy is often required for the induction of remission of autoimmune bullous disorders. In addition, the employment of Intravenous Immunoglobulin (IVIg) and monoclonal antibodies such as rituximab offers alternatives to an immunosuppressive route of treatment. Topical steroids may be used as an adjunctive therapy or in mild or limited cases [3]. The treatment of autoimmune blistering disorders is largely based on clinical experience, as large controlled clinical trials are lacking due to relatively small numbers of patients. A treatment algorithm for bullous pemphigoid and the pemphigus disorders currently does not exist. The aim of this study is to characterize the treatment of autoimmune bullous disease in the United States.Methods
This study was conducted using data available through the National Ambulatory Medical Care Survey (NAMCS). This information is derived from outpatient, non-federally funded physician office visits across the United States and includes approximately 700 million estimated from a sample of 30,000 office visits. Through the use of a multistage probability sample design, the survey tabulates unbiased estimates of physician-patient visits. Each participating office is assigned a time period in which diagnosis codes, prescriptions, procedures, and referrals are recorded at the time of the visit.Examining NAMCS data from 1993 through 2010, patients were included with an ICD-9 diagnosis code of 694.4 (pemphigus vulgaris or pemphigus foliaceus), 694.5 (bullous pemphigoid), or 694.60 or 694.61 (cicatricial pemphigoid with or without ocular involvement). Due to low numbers of subjects, cicatricial pemphigoid was excluded from the analysis. This study focused on the most common autoimmune bullous diseases and did not encompass all of the autoimmune bullous diseases such as linear IgA bullous dermatosis, epidermolysis bullosa acquisita, etc.Demographic information including gender, race, ethnicity, and age of included patients was examined. The most commonly prescribed medications for blistering diseases by both dermatologists and non-dermatologists were also assessed. Visits with multiple diagnoses were excluded when determining leading medications prescribed, to ensure that the medications prescribed were for blistering disease. Through the use of national census data for the year 2000, the national population estimate per 100,000 persons was calculated for sex, race, and ethnicity.Results
From 1993 through 2010, a total of 1,290,000 outpatient visits were recorded for patients treated for autoimmune bullous disorders. Patients were primarily female (570 per 100,000; Table 1), white race (545 per 100,000) and non-Hispanic ethnicity (322 per 100,000). Bullous diseases were most commonly diagnosed in patients aged 70 to 79 (25%; Figure 1). Dermatologists were the treating specialty for 910,000 (71%) visits. Family practitioners (16%), ophthalmologists (7%), and internists (3%) also treated patients with bullous disorders (Figure 2).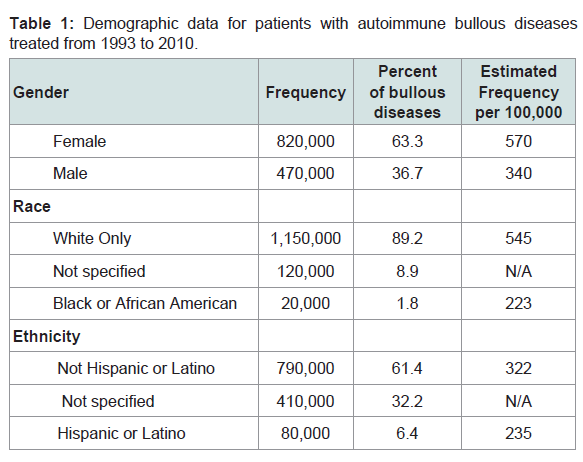
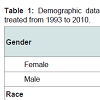
Table 1: Demographic data for patients with autoimmune bullous diseases treated from 1993 to 2010.
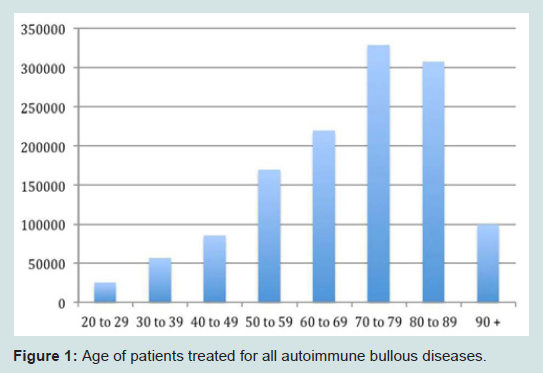
Figure 1: Age of patients treated for all autoimmune bullous diseases.
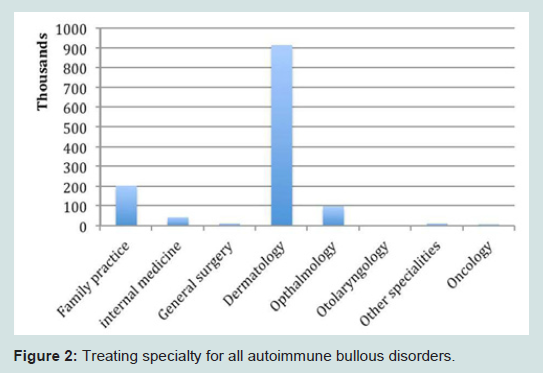
Figure 2: Treating specialty for all autoimmune bullous disorders.
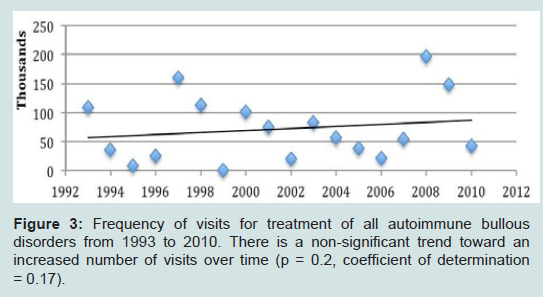
Figure 3: Frequency of visits for treatment of all autoimmune bullous disorders from 1993 to 2010. There is a non-significant trend toward an increased number of visits over time (p = 0.2, coefficient of determination = 0.17).
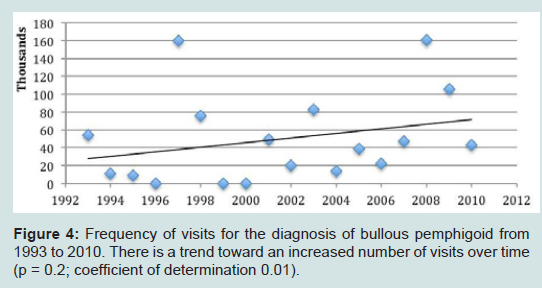
Figure 4: Frequency of visits for the diagnosis of bullous pemphigoid from 1993 to 2010. There is a trend toward an increased number of visits over time (p = 0.2; coefficient of determination 0.01).
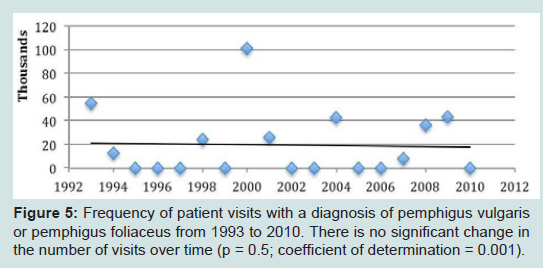
Figure 5: Frequency of patient visits with a diagnosis of pemphigus vulgaris or pemphigus foliaceus from 1993 to 2010. There is no significant change in the number of visits over time (p = 0.5; coefficient of determination = 0.001).
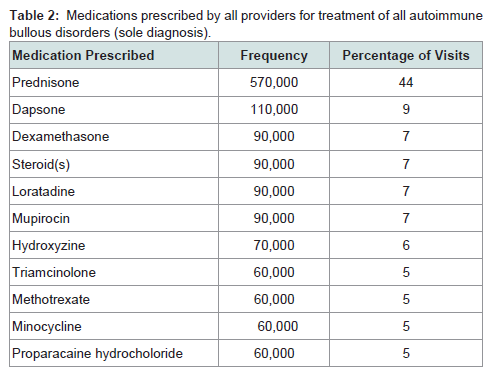
Table 2: Medications prescribed by all providers for treatment of all autoimmune bullous disorders (sole diagnosis).
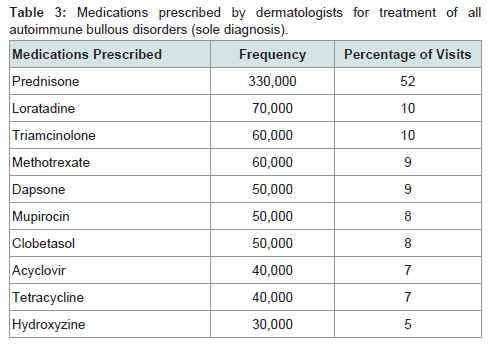
Table 3: Medications prescribed by dermatologists for treatment of all autoimmune bullous disorders (sole diagnosis).
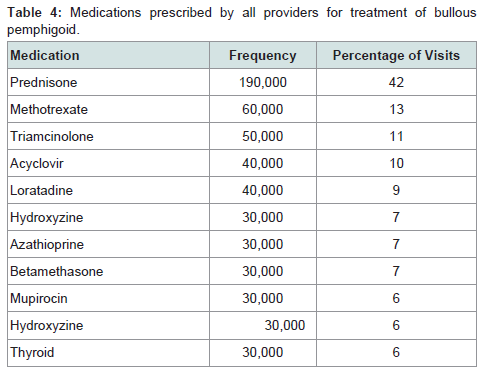
Table 4: Medications prescribed by all providers for treatment of bullous pemphigoid.
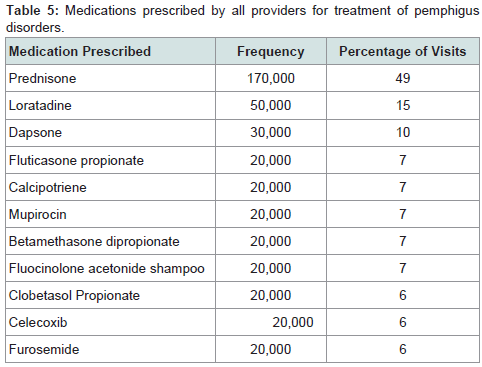
Table 5: Medications prescribed by all providers for treatment of pemphigus disorders.
Discussion
Bullous pemphigoid and pemphigus vulgaris are the prototype autoimmune blistering diseases. Bullous pemphigoid is the most common autoimmune bullous disease and is commonly diagnosed among the elderly population. It is induced by pathogenic autoantibodies which interfere with hemidesmosomal attachment, with resultant subepidermal bulla formation [4]. Clinically, afflicted patients have tense bullae on skin and less frequently mucous membranes. In contrast, pemphigus vulgaris is mediated by autoantibodies directed against desmogleins, key protein components of desmosomes which facilitate cell-cell adhesion in the epidermal and mucosal epithelium [5]. Flaccid bullae rapidly rupture to form non-healing painful erosions, which are slow healing and may be complicated by superinfection. The mortality of autoimmune bullous disorders has decreased dramatically with appropriate treatment, which typically includes oral corticosteroids and other systemic immunomodulating agents aimed at suppressing the aberrant autoimmune response. Adequate control of autoimmune bullous diseases often requires long-term immunosuppression for months to years. However, immune modulating therapies are associated with significant potential adverse effects such as serious infections, which may be increased in elderly patients [6].The diagnosis of autoimmune blistering disorders is aided by histopathologic examination as well as direct and indirect immunofluorescence microscopy [11,7,8]. Hence, many patients presenting with bullous lesions are initially evaluated by a dermatologist, often in conjunction with a dermatopathologist. However, ophthalmologists also may make a diagnosis of an autoimmune bullous disease particularly when conjunctival mucosa is involved [9]. Novel diagnostic approaches examining antigen or antibody levels in serum or tissue are becoming increasingly available.Numerous treatments have been reported as effective therapies for the treatment of bullous pemphigoid. No consensus exists regarding ‘standard’ therapy as providers balance efficacy and side effects of potential therapies. The aim of this study is to examine the mostly commonly prescribed medications for the treatment of bullous pemphigoid in the United States and trends over time regarding therapy to better characterize current prescribing habits.From 1993 to 2010 the number of patients treated for bullous pemphigoid and pemphigus has not dramatically changed. This may imply that the incidence of autoimmune blistering diseases has remained relatively stable in recent years. In addition, patients with bullous pemphigoid and pemphigus were often older which concurs with results from other studies [10]. Data for cicatricial pemphigoid was also examined; however, the number of patients was too small to draw conclusions for this patient population.>Consistent with previous studies, oral corticosteroids are considered first line therapy for the treatment of bullous pemphigoid. The study highlights the wide variations in other treatments prescribed for bullous pemphigoid. Various systemic immune modulating agents have been reported as effective adjunctive therapy including azathioprine, mycophenolate mofetil, cyclophosphamide, methotrexate, cyclosporine, and intravenous immune globulin (IVIG) [2,11]. Rituximab, a monoclonal antibody directed against the B-cell surface marker CD20, is also used in the treatment of pemphigus [12]. Our data reveals that dermatologists prescribe prednisone to almost half of patients with autoimmune bullous disorders. The need for aggressive treatment of pemphigus vulgaris is evidenced by its previously very high mortality rate, prior to the advent of oral corticosteroids. Unfortunately, systemic immunosuppressive agents are not without potential morbidity and mortality, which may be increased in elderly patients who are relatively immunosuppressed compared to healthy adults [13]. In order to stave off potential sequelae associated with chronic systemic corticosteroid therapy, alternative immune modulating agents are often added to treatment. These agents may decrease the required dose or duration of corticosteroid therapy while maintaining disease control. Although prednisone is the most commonly prescribed immunosuppressive agent, there are currently no consensus guidelines or recommended therapeutic ladder for addition of a steroid-sparing immunosuppressive agent. Further studies are needed to determine the immunosuppressive agent with the optimal risk-benefit profile for treatment of autoimmune bullous disorders, particularly in elderly patients.The high frequency of prescription of non-sedating oral antihistamine medications such as loratadine for the treatment of autoimmune bullous disorders observed in this study is somewhat curious. These medications would not be expected to alter the course of bullous diseases, but may have been prescribed for symptomatic relief. Bullous pemphigoid, especially in early or urticarial phases, may be associated with severe itching. Although we attempted to exclude medications prescribed for other disorders by examining only patients with a sole diagnosis of an autoimmune bullous disorder, it is possible that antihistamine medications were prescribed for an unrelated condition.Although the data indicates that patients with a sole diagnosis of autoimmune blistering disease were taking other nonimmunosuppressive drugs such as Acyclovir, Furosemide, Celecoxib, etc., for the management of their bullous disease, it may represent other concomitant unlisted confounding diagnoses and not treatment for bullous disease.This study is observational and relies upon extrapolation of deidentified data based on physician-patient outpatient encounters. Inaccurate coding of patients with autoimmune bullous disorders may have resulted in inclusion of patients with other diseases or exclusion of some patients with the examined diagnoses.References
- Schmidt E, Torre R.D, Borradori L (2011) Clinical Features and Practical Diagnosis of Bullous Pemphigoid. Dermatol Clin 29: 427-438.
- Kneisel A, Hertl M (2011) Autoimmune bullous skin diseases. Part 2: Diagnosis and therapy. J Dtsch Dermatol Ges 9: 927-947.
- Yancey KB, Egan CA (2000) Pemphigoid: Clinical, Histologic, Immunopathologic, and Therapeutic considerations. JAMA 284: 350-356.
- Zillikens D, Giudice GJ (1999) BP180/type XVII collagen: its role in acquired and inherited disorders or the dermal-epidermal junction. Arch Dermatol Res 291: 187-194.
- Karpati S, Amagai M, Prussick R, Cehrs K, Stanley JR (1993) Pemphigus vulgaris antigen, a desmoglein type of cadherin, is located within keratinocyte desmosomes. J Cell Biol 122: 409-415.
- Fine JD (2012) Bullous diseases. In: Moschella SL, Hurley HJ, eds. Dermatology, Vol 3, Philadelphia, PA: WB Saunders Co.
- Di Zenzo G, Marazza G, Borradori L (2007) Bullous pemphigoid: physiopathology, clinical features and management. Adv Dermatol 23: 257-288.
- Gammon WR, Kowalewski C, Chorzelski TP, Kumar V, Briggaman RA, et al. (1990) Direct immunofluorescence studies of sodium-chloride-separated skin in the differential diagnosis of bullous pemphigoid and epidermolysis bullosa acquisita. J Am Acad Dermatol 22: 664-670.
- Kirzhner M, Jakobiec FA (2011) Ocular cicatricial pemphigoid: a review of clinical features, immunopathology, differential diagnosis, and current management. Semin Ophthalmol 26: 270-277.
- Parker SR, MacKelfresh J (2011) Autoimmune blistering diseases in the elderly. Clin Dermatol 29: 69-79.
- Bystryn JC, Steinman NM (1996) The adjuvant therapy of pemphigus. An update. Arch Dermatol 132: 203-212.
- El Tal AK, Posner MR, Spigelman Z, Ahmed AR (2006) Rituximab: a monoclonal antibody to CD20 used in the treatment of pemphigus vulgaris. J Am Acad Dermatol 55: 449-459.
- Da Silva AV, Valones MA, Guimaraes RP, de Castro JF, Carvalho Ade A (2008) Pemphigus vulgaris: a therapeutic option for disease control. Gen Dent 56: 700-703.