
Journal of Clinical and Investigative Dermatology
Download PDF
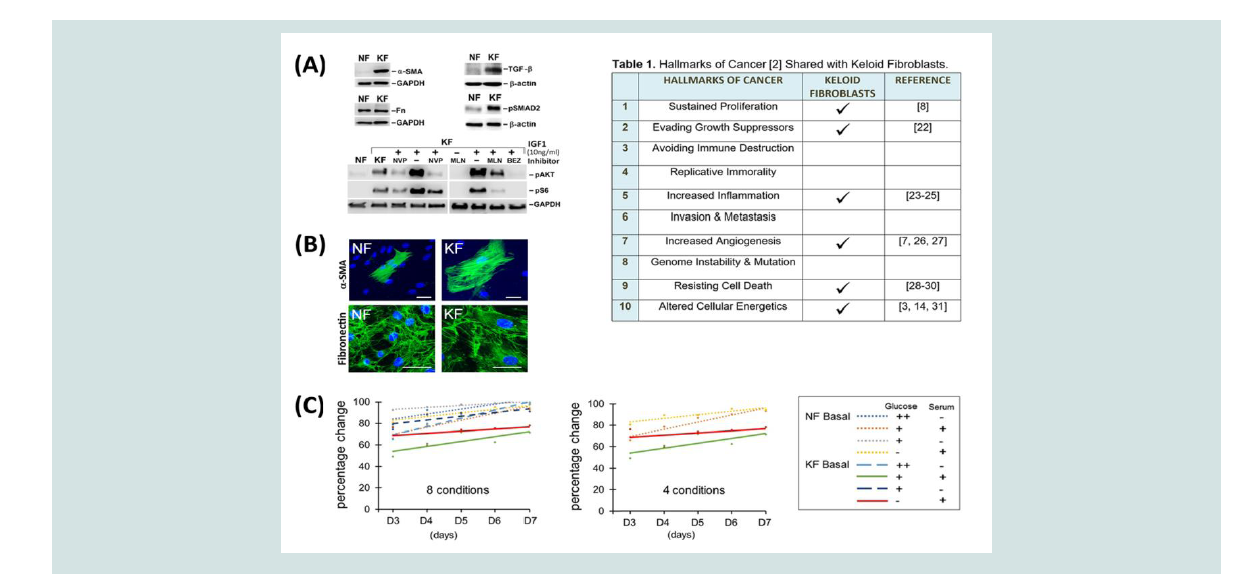 Figure 1: Keloid fibroblast characteristics and viability. (A) Western analysis of Keloid Fibroblasts (KF) protein expression and signaling pathway activation
compared with control, Normal Fibroblasts (NF). Elevated expression of α-SMA, fibronectin, TGFβ and pSMAD2 in unstimulated KFs. Analysis of IGF1 (10 ng/ml)
signaling in KF and NF demonstrates activation of pAKT, pS6 at 30 min after stimulation. For KF, these kinases were activated at the basal state. Inhibitors (NVP,
1µM NVPAEW541; MLN, 3µM MLN1117; BEZ, 100nM BEZ235) were added 30 min before IGF1 stimulation in lanes as indicated. Twenty microgram of cell lysate
was loaded in each lane. GAPDH or β-actin were used to show equal loading. (B) Immunofluorescent staining of (keloid fibroblasts), using α-SMA, fibronectin, or
phalloidin as indicated; nuclei were stained with DAPI in blue. Scale bar, 100 µm. (C) Percentage change in viability plotted for conditions specified in the legend.
Glucose (++, 4.5g/L; +, 1g/L) and/or serum (+, present; -, absent) changes from basal, complete media are indicated. These changes did not reach statistical
significance (p < 0.05) using ANCOVA; 4 conditions showing the greatest difference are also indicated (middle panel).
Figure 1: Keloid fibroblast characteristics and viability. (A) Western analysis of Keloid Fibroblasts (KF) protein expression and signaling pathway activation
compared with control, Normal Fibroblasts (NF). Elevated expression of α-SMA, fibronectin, TGFβ and pSMAD2 in unstimulated KFs. Analysis of IGF1 (10 ng/ml)
signaling in KF and NF demonstrates activation of pAKT, pS6 at 30 min after stimulation. For KF, these kinases were activated at the basal state. Inhibitors (NVP,
1µM NVPAEW541; MLN, 3µM MLN1117; BEZ, 100nM BEZ235) were added 30 min before IGF1 stimulation in lanes as indicated. Twenty microgram of cell lysate
was loaded in each lane. GAPDH or β-actin were used to show equal loading. (B) Immunofluorescent staining of (keloid fibroblasts), using α-SMA, fibronectin, or
phalloidin as indicated; nuclei were stained with DAPI in blue. Scale bar, 100 µm. (C) Percentage change in viability plotted for conditions specified in the legend.
Glucose (++, 4.5g/L; +, 1g/L) and/or serum (+, present; -, absent) changes from basal, complete media are indicated. These changes did not reach statistical
significance (p < 0.05) using ANCOVA; 4 conditions showing the greatest difference are also indicated (middle panel).
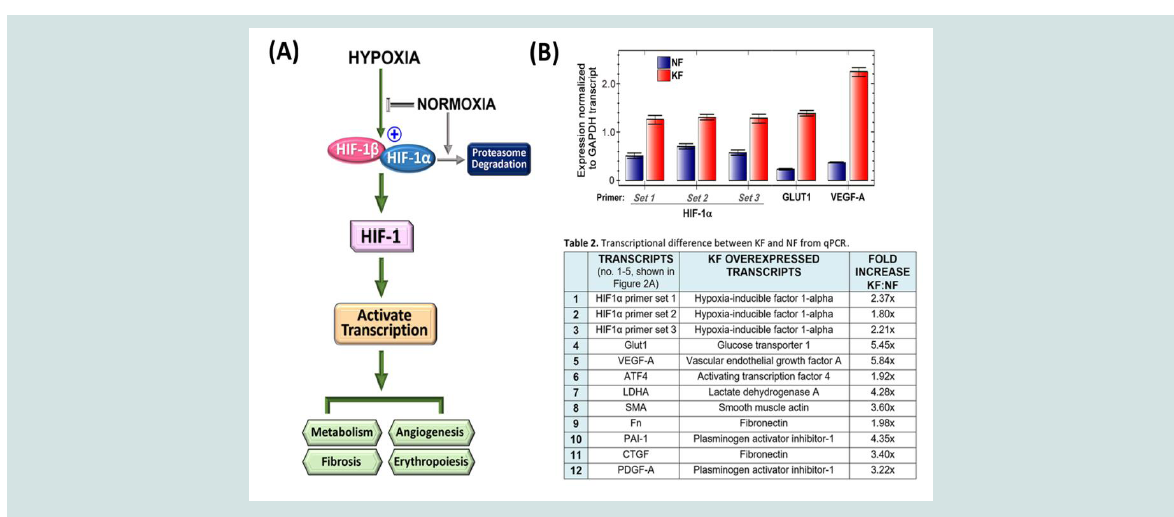 Figure 2: The HIF-1 pathway is activated in unstimulated keloid fibroblasts, affecting metabolism and angiogenesis. (A) The hypoxia pathway flowchart demonstrates
activation of the HIF-1 transcription factor when the HIF-1a subunit is available to bind HIF-1b. Under normal oxygen levels, HIF-1a is synthesized as a protein,
but is degraded before interaction with the HIF- 1b subunit. (B) qPCR analysis of transcript levels for HIF-1a and HIF-1 regulated genes, GLUT1 and VEGF-A, in
nstimulated NF or KF normalized to the expression of GAPDH. Three sets of HIF-1a primers were used to ensure accurate estimation.
Figure 2: The HIF-1 pathway is activated in unstimulated keloid fibroblasts, affecting metabolism and angiogenesis. (A) The hypoxia pathway flowchart demonstrates
activation of the HIF-1 transcription factor when the HIF-1a subunit is available to bind HIF-1b. Under normal oxygen levels, HIF-1a is synthesized as a protein,
but is degraded before interaction with the HIF- 1b subunit. (B) qPCR analysis of transcript levels for HIF-1a and HIF-1 regulated genes, GLUT1 and VEGF-A, in
nstimulated NF or KF normalized to the expression of GAPDH. Three sets of HIF-1a primers were used to ensure accurate estimation.
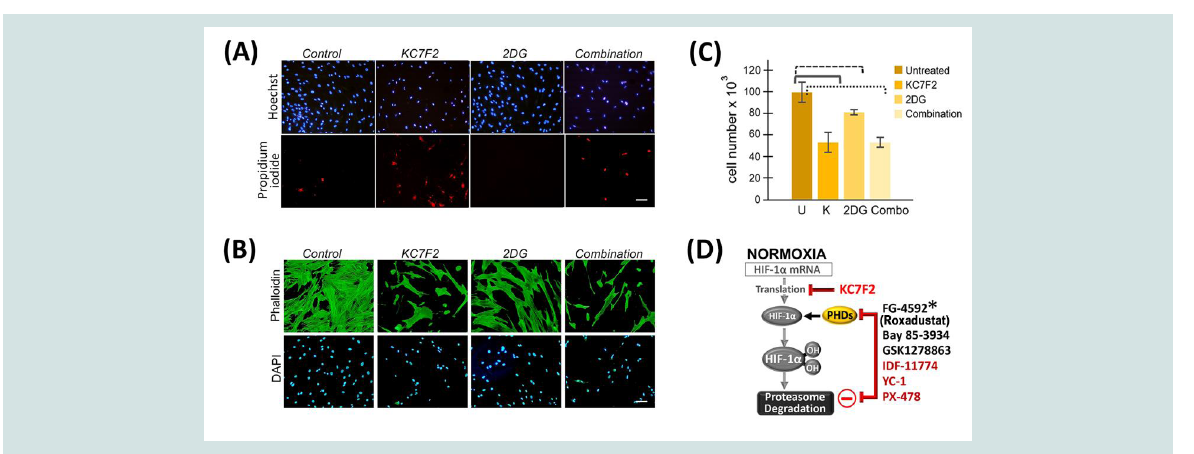 Figure 3: Keloid fibroblasts are more efficiently targeted by HIF1α inhibition. (A, B) KFs were untreated, or treated with 40 µM KC7F2, and/or 10mM 2DG. (A)
Vital staining of KFs using Hoescht 33342 to label nuclei (top) and propidium iodide to detect dying cells (bottom). (B) The same treatment was performed again
where cells were fixed and stained with phalloidin (top) and DAPI (bottom). (C) Nuclei counts are presented. A pairwise T-test demonstrated statistical significance
between untreated control KFs (U) and all other conditions (p ≤ 0.05); against KC7F2 alone (K, at p = 0.05); against 2-DG (p = 0.04); or against the combination
(p = 0.002). (D) The transcriptional activity of HIF-1 can be modified by chemical compounds that interfere with the expression and processing of HIF-1α. Prolyl
hydroxylation of HIF-1α targets it for degradation. Prolyl Hydroxylase Domain (PHD) proteins can be inhibited by small molecule inhibitors such as FG-4592
(*Roxadustat, approved in China for anemia treatment), BAY 85-3934 or GSK1278863. Additional inhibitors such as KC7F2, YC-1, IDF-11774, and PX-478 reduce
HIF-1α protein levels.
Figure 3: Keloid fibroblasts are more efficiently targeted by HIF1α inhibition. (A, B) KFs were untreated, or treated with 40 µM KC7F2, and/or 10mM 2DG. (A)
Vital staining of KFs using Hoescht 33342 to label nuclei (top) and propidium iodide to detect dying cells (bottom). (B) The same treatment was performed again
where cells were fixed and stained with phalloidin (top) and DAPI (bottom). (C) Nuclei counts are presented. A pairwise T-test demonstrated statistical significance
between untreated control KFs (U) and all other conditions (p ≤ 0.05); against KC7F2 alone (K, at p = 0.05); against 2-DG (p = 0.04); or against the combination
(p = 0.002). (D) The transcriptional activity of HIF-1 can be modified by chemical compounds that interfere with the expression and processing of HIF-1α. Prolyl
hydroxylation of HIF-1α targets it for degradation. Prolyl Hydroxylase Domain (PHD) proteins can be inhibited by small molecule inhibitors such as FG-4592
(*Roxadustat, approved in China for anemia treatment), BAY 85-3934 or GSK1278863. Additional inhibitors such as KC7F2, YC-1, IDF-11774, and PX-478 reduce
HIF-1α protein levels.
Research Article
Targeting Keloid Fibroblasts by Inhibition of Hypoxia Signaling
Richert-Jones J1, Mantel A1, Ricks-Santi L2, Harvey V1 and Chan J1*
1Hampton University Skin of Color Research Institute, USA
2Hampton University Cancer Research Center, USA
*Address for Correspondence: Joanne Chan, Scientific Director and Associate Professor, Hampton University Skin of Color Research Institute (HUSCRI) 100 William R. Harvey Way, Hampton, VA 23668, Hampton, VA 23668, USA, Tel: 757-726-6058; E-mail: joanne@chanlab.org
Submission: 26 August, 2020;
Accepted: 28 September, 2020;
Published: 30 September, 2020
Copyright: © 2020 Richert-Jones J, et al. This is an open access article
distributed under the Creative Commons Attribution License, which permits
unrestricted use, distribution, and reproduction in any medium, provided the
original work is properly cited.
Abstract
Keloids are persistent raised scars that are difficult to treat because attempts
at removal typically results in recurrence. Keloid fibroblasts are the abnormal
cell type responsible for the continuous scar protein deposition in this fibrotic
skin disease that involves enhanced TGFβ activity. An effective therapy that can
eliminate keloid fibroblasts and promote normal healing is needed. We examined
the cellular and molecular differences between keloid and normal skin fibroblasts
to identify characteristics that may be targeted for therapy. By limiting serum and/
or glucose availability, we found that keloid fibroblasts are sensitive to glucose levels
but not to serum withdrawal. Treatment with 2- deoxyglucose, a preclinical drug
that blocks glycolytic metabolism, can reduce keloid fibroblast cell size. However,
recovery occurs upon drug removal, indicating a cytostatic effect. To eliminate
keloids, it would be necessary to induce cell death in order to disrupt the cycle of
continuous fibrosis. Thus, we examined the role of HIF-1, a central transcription
factor that regulates both glycolytic metabolism and fibrosis, to determine whether
blocking its activity in keloid fibroblasts could yield a cytotoxic outcome. Using
a chemical inhibitor against HIF-1, we observed a significant reduction in keloid
fibroblast numbers. Although small molecule HIF-1 inhibitors have been under
development for anemia and cancer therapy, their role in regulating fibrotic genes
has led researchers to consider their potential use in the treatment of lung or kidney
fibrosis. Since keloid disease may be considered a form of chronic skin fibrosis,
reducing HIF-1 activity could provide a therapeutic strategy for keloid treatment.
Keywords
Keloid; Abnormal fibroblasts; Metabolic reprogramming; Hypoxia; Glycolysis
Abbreviations
KF: Keloid Fibroblast; NF: Normal Fibroblast; HIF-1: Hypoxia Responsive Factor-1; 2-DG: 2-Deoxyglucose; qPCR: quantitative Polymerase Chain Reaction; TGFβ: Transforming Growth Factorbeta; mTORC1: mechanistic Target of Rapamycin Complex 1; ANCOVA: Analysis of Co-Variance
Introduction
Keloids are dense, enlarged scars that extend beyond the borders
of the original injury. They are prevalent in skin of color individuals
and are difficult to treat [1] because removal generates another
injury and the recurrence of an even larger keloid. Since they only
occur in humans, animal models have not been sufficient to study
this disease [1]. The keloid fibroblast (KF) is the abnormal cell type
responsible for producing and depositing an excessive amount
of matrix proteins into the scar. Research with KFs has identified
enhanced TGFβ signaling, as well as features shared with cancer
cells [2], such as a dependence on glucose for energy metabolism
[3], elevated mTORC1 activation [4,5], and increased transcriptional
activity of HIF-1 (Hypoxic Response Factor-1; [6,7]). In this study,
we determined cell viability under conditions of limited glucose and/
or serum [8], or upon treatment with inhibitors of glycolysis and/or
hypoxia signaling. We found that treatment with a HIF-1α inhibitor
effectively reduced KF viability, suggesting that targeting the hypoxia
signaling pathway could provide a novel strategy that can eliminate
pathological KF cells and promote normal healing of keloid scars.
Materials and Methods
Fibroblast assays:
Keloid fibroblasts (CRL1762) and race‐matched normal
fibroblasts (CRL2439) were purchased from the American Type
Culture Collection (ATCC; 217 Perry Parkway Gaithersburg, MD
20877, USA) and cultured in DMEM (Gibco cat. no. 1995-065;
Life Technologies Limited 3 Fountain Drive Paisley, PA49RF, UK)
containing 10% FBS, 1:500 amphotericin B, and 1:100 penicillin/
streptomycin (Gibco cat. no. 26140-079, 15290-026, 15140-122;
Life Technologies Corporation 3175 Staley Road Grand Island,
NY 14072, USA) as described [9]. Cell viability was determined by
hemocytometry and Trypan Blue dye exclusion. Equal cell numbers
were plated at day 0, then treated under 8 distinct conditions.
Chemical inhibitors used were: 2-deoxyglucose (2-DG; SigmaAldrich cat. no. D6134; 3050 Spruce Street St. Louis, MO 63103,
USA) at 10 mM for 2-16 h, or 40 µM KC2F7 for 16 h, NVP-AEW541,
MLN1117 and BEZ235 (Selleck Chemicals cat. no. S7946; 14408;
no. S1034, S8581, S1009; W Sylvanfield Drive Houston, TX 77014,
USA). For immunofluorescence, cells were fixed, then permeabilized
and stained with an anti-SMA or anti-fibronectin antibody (SigmaAldrich/MilliporeSigma, cat. No. A5228, MAB1926; 400 Summit
Drive, Burlington, MA 1803, USA), Alexa Fluor 594 phalloidin,
Alexa Fluor 488 phalloidin, Hoechst-33342 (Life Technologies cat.
no. A12349, A12381, H1399; 29851 Willow Creek Road Eugene, OR
97402, USA), and/or DAPI (Molecular Probes, Inc. cat. no. 62248;
3747 N Meridian Road Rockford, IL 61101, USA). For live imaging,
propidium iodide (BioSure, Inc. cat. no. 1032; 1050 Whispering Pines
Lane, STE F, Grass Valley, CA 95945, USA) is used to stain dying
cells and Hoechst-33342 is used to stain nuclei. Western blot analysis
was performed as previously described [9]. Twenty micrograms of
cell lysates per lane was used in standard western analysis. Blots
were incubated with the indicated primary antibodies at 4 °
C,
overnight, the appropriate secondary horseradish peroxidase (HRP)-
conjugated IgG (Life Technologies) used at 1:4000 in TBST for 1 h
at RT. Immunoreactive protein bands were detected using Western blot luminol reagent (Bio-Rad cat. no. P10026378/ P10026379, 1000
Alfred Nobel Drive Hercules, CA, USA). Primary antibodies were:
phospho-SMAD2 (pSMAD2) and phospho-S6 (pS6) (Cell Signaling
Technology, Inc. cat. no. 18338T and no. 4857S, respectively; 3 Trask
Lane, Danvers, MA 01923, USA).
Statistical analysis:
The results were expressed as the mean ± Standard Deviation
(SD). Statistical analyses were performed using the SPSS 22.0
software, one-way ANCOVA for group comparisons or a paired
T-test for pairwise comparisons. A value of p ≤ 0.05 was considered
statistically significant.
Results and Discussion
Keloids are often compared with cancers in terms of their chronic
and persistent growth. Some of these features are preserved in keloid
fibroblasts (KFs); therefore, with the absence of an animal model,
fibroblasts derived from keloid scars provide an important cellbased model that retains key features of keloids. Of the 10 hallmarks of cancer, keloids share 6 of them [2] (Table 1). To validate some of these characteristics [1], we used western and immunofluorescent analyses to examine the expression of relevant proteins and activated
pathways in keloid fibroblasts (KFs), but not in normal skin fibroblasts
(NFs; Figure 1A). KFs typically express high levels of Smooth Muscle
Actin (SMA), a recognized property of the transitional myofibroblast
[10]. As reported by others [11], we also noted expression of TGF-β
and detection of phospho-SMAD2 (pSMAD2) indicating chronic
pathway activation in unstimulated KFs. In addition, mTORC1 (mammalian or mechanistic target of rapamycin complex 1) activity is
also elevated in KFs, as shown by phospho-S6 (pS6) detection (Figure 1A). To demonstrate specificity of these activated signals, we also used
IGF1 stimulation, either alone or in the presence of highly selective
chemical inhibitors to block the signaling pathway at several nodes:
IGF1 receptor (using NVP-AEW541), PI3Kα (phosphoinositide
3-kinase-alpha, using the PI3Kα inhibitor MLN1117), or both PI3K
and mTORC1 (using the dual inhibitor NVP-BEZ235 [12,13]). In
addition, we also observed an enlargement in KF cell size, in accord
with enhanced mTORC1 activity, shown by immunofluorescent
staining of SMA [5] (Figure 1B). Although the expression of some
proteins is highly elevated, others are not. For example, fibronectin
expression is similar in both types of fibroblasts (Figure 1A and 1B).
Previous studies have shown a preference for glycolytic metabolism in KFs, where chemical inhibitors against this pathway were used to evaluate their potential use in keloid therapy [3,14]. In a comprehensive study by Vincent et al. (2008); [3]) changes in lactate or ATP production were reduced when glycolysis was impaired by 3
structurally distinct inhibitors. In another study, Li et al. (2018; [14]),
2-Deoxyglucose (2-DG) was shown to reduce KF viability over 4 days.
Although both studies suggested that chemical glycolysis blockade
could be targeted in treating keloids, it is not known whether KFs can
be selectively inhibited while allowing NFs to survive.
To examine this further, we investigated whether limiting glucose
availability could reveal a difference in viability between NF and KF.
Thus, we determined live cell numbers when these fibroblasts were
grown in media with reduced glucose and/or serum, as compared with complete media. For each type of fibroblast, at least 300,000 cells per
flask were seeded in triplicate on day 0. Cell numbers were counted
from days 3-7 using hemocytometry (Figure 1C). To highlight
glucose-specific effects, we presented a simplified graph to show that
both NF and KF cells were affected at similar rates when glucose
levels are reduced to 22% or 0%. Thus, limiting glucose availability or
antagonizing enzymes in the glycolysis pathway may not provide an
effective means for keloid treatment. This observation was confirmed
using an Analysis of Co-Variance (ANCOVA), to show that lowering
glucose and/or serum levels did not yield statistical differences
between cells (p > 0.05; Figure 1C).
We also examined whether preclinical drugs antagonizing
overactivated pathways in KFs could destroy this abnormal cell type
(Figure 1, data not shown). These include 2- DG to inhibit glycolysis,
MLN-1117 to inhibit PI3K, BEZ235 to antagonize both PI3K and
mTORC1, as well as KC7F2 (inhibiting HIF-1α; [15]). We reasoned
that antagonizing the HIF-1 signaling pathway would limit the
expression of genes for both glycolysis and fibrosis, which would
be beneficial in keloid treatment (Figure 2A). HIF-1 targeting has
also been considered for the treatment of muscle and organ fibrosis
[16-18]. However, KFs are cultured under normoxic conditions;
therefore, an abnormal activation of the hypoxia signaling pathway
needs to be present in order for inhibitors to work. Elevated HIF-1
activity has been reported in KFs. In the Vincent et al. (2008) study,
overactivation of hypoxia signaling in KFs was confirmed as these
cells were still able to generate ATP in the presence of 2 hypoxia
mimics: desferrioxamine and cobalt chloride [3]. We examined HIF1α transcript levels using qPCR to find an ~2-fold increase. Using 3
distinct sets of primers, we confirmed HIF-1α overexpression in KFs
at approximately ~2-fold, with a range of 1.6 to 2.3-fold, compared
with NFs. Although HIF-1α mRNA was not highly overexpressed,
HIF-1 regulated genes, GLUT1 (glucose transporter-1) and VEGF-A
(vascular endothelial growth factor-A) transcripts, were increased in excess of 5-fold (Figure 2 and Table 1; see supplemental information).
Thus, using protein or transcript detection, we have demonstrated
increased TGF-β, GLUT1 and VEGF levels in the basal, unstimulated
state in KFs. Since overactivation of these pathways can impact
fibrosis, metabolism and angiogenesis, antagonizing HIF-1 function
could affect several pathways that are abnormally activated in KFs
(Figure 2A,2B).
To determine whether a combined blockade of HIF-1α and
glycolysis might be effective in eradicating KFs, we used a HIF-1α
inhibitor, KC7F2 [15], either alone or in combination with 2-DG,
on KF viability. Within 24 h of treatment, KFs were eliminated.
Therefore, we refined conditions to obtain ~ 50% reduction in KF
viability (Figure 3A-3C). When 2-DG was present alone, cell viability
was only reduced to 80%. In the combined treatment, KF cell numbers
remained similar to ~ 50%, as observed with KC7F2 treatment alone,
suggesting that 2-DG did not contribute to enhancing KF elimination
(Figure 3C). Furthermore, vital staining showed that increased cell
death, indicated by propidium iodide, occurred in the presence of the
KC7F2 HIF-1α inhibitor (Figure 3A). In addition, immunofluorescent
analysis demonstrated severe alterations in cell morphology in cells
treated with the HIF-1α inhibitor but not with 2-DG (Figure 3B).
Thus, in contrast to previous studies [3,14], our data demonstrated
that blocking glycolysis does not lead to the elimination of the
abnormal KF cells, an important goal for any keloid therapy that aims
to reduce recurrence and promote healing.
HIF-1 is a master regulator that controls essential processes
including metabolism, fibrosis, angiogenesis and erythropoiesis
(Figures 2A and 3D). Small molecule inhibitors that alter the
functional levels of the HIF-1α subunit have been in preclinical
and clinical development for cancer treatment and anemia [19,20].
Currently, one of the HIF prolyl hydroxylase inhibitors, FG-4592
(Roxadustat, clinically approved in China in December 2018), has been successful in clinical phase II and III trials for treating anemia in
patients with chronic kidney disease [21]. As more HIF-1α inhibitors
become clinically approved, their additional use for keloid disease
could be accelerated based on clinical safety data. For keloid disease
in particular, these inhibitors can be locally administered through an
intralesional injection. Thus, direct targeting of KFs may be achieved
without inducing an additional skin injury.
Conclusion
Our study provides a novel drug target and treatment strategy
for patients with keloid disease. This method may be extended to
other skin disorders such as localized scleroderma, hidradenitis
suppurativa, nephrogenic fibrosing dermopathy, and eosinophilic
fasciitis, where localized fibrotic lesions persist.
Data Availability
Data presented in this manuscript is available to the research
community without restriction at the time of online publication.
References
Citation
Richert-Jones J, Mantel A, Ricks-Santi L, Harvey VM, Chan J. Targeting Keloid Fibroblasts by Inhibition of Hypoxia Signaling. J Clin Investigat
Dermatol. 2020;8(2): 5