
Journal of Clinical & Medical Case Reports
Download PDF
Case Report
*Address for Correspondence: Derrick C. Wan, MD, Assistant Professor, Division of Plastic and Reconstructive Surgery, Department of Surgery, Stanford University School of Medicine, 257 Campus Drive West, Stanford, CA, 94305-5148, USA, Tel: 650) 723-7073; Fax: (650) 736-1705; E-mail: dwan@stanford.edu
Citation: Wright EJ, Garza RM, Wan DC. Hereditary Sensory and Autonomic Neuropathy: Case Report and Discussion. J Clin Med Case Reports. 2013;1(1): 4.
Copyright © 2013 Derrick C. Wan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN 2332-4120 | Volume: 1, Issue: 1
Submission: 08 April 2013| Accepted: 18| June 2013| Published: 24 Jun 2013
 Initial differential diagnosis included herpetic whitlow with associated Herpes Simplex Virus (HSV) infection, gingivostomatitis, ecthyma, bullous impetigo, pyoderma gangrenosum, vasculitis, spider bite, or anthrax. The patient was placed in isolation and started on intravenous antibiotics and acyclovir empirically. A HSV/varicella zoster virus direct fluorescent antibody test was performed on the thumb and tongue lesions were both negative, as were wound cultures and anthrax PCR tests. The lesions were managed with local wound care, and auto-delamination of the eschar with resolution of cellulitis occurred over the following two days. The patient was discharged on a course of oral clindamycin.
Initial differential diagnosis included herpetic whitlow with associated Herpes Simplex Virus (HSV) infection, gingivostomatitis, ecthyma, bullous impetigo, pyoderma gangrenosum, vasculitis, spider bite, or anthrax. The patient was placed in isolation and started on intravenous antibiotics and acyclovir empirically. A HSV/varicella zoster virus direct fluorescent antibody test was performed on the thumb and tongue lesions were both negative, as were wound cultures and anthrax PCR tests. The lesions were managed with local wound care, and auto-delamination of the eschar with resolution of cellulitis occurred over the following two days. The patient was discharged on a course of oral clindamycin.
 HSAN-I is a slowly progressive autosomal dominant disorder typically presenting between the second and fifth decades of life [5]. Patients with HSAN-I suffer from loss of pain and temperature sensation beginning in the lower limbs and spreading proximally. Symptoms may be associated with lancinating pain and ulcerations which ultimately lead to a need for limb amputation [6]. When present, autonomic disturbances manifest as hypohydrosis. Multiple genes have been associated with the HSAN-I phenotype including missense mutations in serine palmitoyltransferase (SPTLC1, SPTLC2), atlastin-1 (ATL1), and DNA methyltransferase 1 (DNMT1). Serine palmitoyltransferase catalyzes the formation of sphingolipids which are important structures in the plasma membrane enabling efficient signal transduction [7, 8]. Abnormal enzyme function has also been shown to result in accumulation of aberrant sphingolipid metabolites such as 1-deoxysphinganine which is toxic to primary sensory neurons [9]. In one patient, HSAN-I was also found to be associated with a missense mutation in ATL1, an atlastin GTPase family member [10]. Depletion of ATL1 in rat cortical neurons has been reported to inhibit axonal elongation, and in other patients with neurodegenerative diseases, ATL1 mutations have been linked to degeneration of the upper motor neurons, causing weakness and lower limb spasticity, along with peripheral neuropathy [11, 12]. Finally, heterozygous missense mutations in DNMT1 have been reported in four patients with HSAN-I, dementia, and hearing loss [13]. DNMT1 catalyzes direct methyl group transfer from S-adenosyl methionine to DNA, and in those with impaired DNMT1 function, global hypomethylation has been observed [3]. How this results in progressive sensory loss has yet to be determined, but nonetheless highlights a potential role for epigenetic modifications in the development of neurodegenerative conditions [14].
HSAN-I is a slowly progressive autosomal dominant disorder typically presenting between the second and fifth decades of life [5]. Patients with HSAN-I suffer from loss of pain and temperature sensation beginning in the lower limbs and spreading proximally. Symptoms may be associated with lancinating pain and ulcerations which ultimately lead to a need for limb amputation [6]. When present, autonomic disturbances manifest as hypohydrosis. Multiple genes have been associated with the HSAN-I phenotype including missense mutations in serine palmitoyltransferase (SPTLC1, SPTLC2), atlastin-1 (ATL1), and DNA methyltransferase 1 (DNMT1). Serine palmitoyltransferase catalyzes the formation of sphingolipids which are important structures in the plasma membrane enabling efficient signal transduction [7, 8]. Abnormal enzyme function has also been shown to result in accumulation of aberrant sphingolipid metabolites such as 1-deoxysphinganine which is toxic to primary sensory neurons [9]. In one patient, HSAN-I was also found to be associated with a missense mutation in ATL1, an atlastin GTPase family member [10]. Depletion of ATL1 in rat cortical neurons has been reported to inhibit axonal elongation, and in other patients with neurodegenerative diseases, ATL1 mutations have been linked to degeneration of the upper motor neurons, causing weakness and lower limb spasticity, along with peripheral neuropathy [11, 12]. Finally, heterozygous missense mutations in DNMT1 have been reported in four patients with HSAN-I, dementia, and hearing loss [13]. DNMT1 catalyzes direct methyl group transfer from S-adenosyl methionine to DNA, and in those with impaired DNMT1 function, global hypomethylation has been observed [3]. How this results in progressive sensory loss has yet to be determined, but nonetheless highlights a potential role for epigenetic modifications in the development of neurodegenerative conditions [14].
 HSAN-III is also known as familial dysautonomia or Riley-Day syndrome. This subtype shows autosomal recessive inheritance and occurs almost solely in Eastern European Jews. Manifesting at birth, widespread autonomic disturbances are noted along with sensory neuropathy [3]. In addition to decreased pain and temperature perception, patients also suffer from impaired coordination from cerebellar atrophy, decreased taste, alacrima, and hyperhidrosis [20]. Of all HSAN subtypes, patients with HSAN-III have the shortest life expectancy [3]. Only mutations in elongator complex protein 1, also known as IκB kinase complex-associated protein (IKBKAP), have been associated with HSAN-III [21]. This protein is required for RNA polymerase II complex assembly, and gene knockdown has been reported to inhibit peripheral neurogenesis, neuronal differentiation and development, and cerebral oligodendrocyte dyelination [22]. Homozygous IKBKAP knockout mice die in utero with abnormalities noted in both vascular and neural development [23].
HSAN-III is also known as familial dysautonomia or Riley-Day syndrome. This subtype shows autosomal recessive inheritance and occurs almost solely in Eastern European Jews. Manifesting at birth, widespread autonomic disturbances are noted along with sensory neuropathy [3]. In addition to decreased pain and temperature perception, patients also suffer from impaired coordination from cerebellar atrophy, decreased taste, alacrima, and hyperhidrosis [20]. Of all HSAN subtypes, patients with HSAN-III have the shortest life expectancy [3]. Only mutations in elongator complex protein 1, also known as IκB kinase complex-associated protein (IKBKAP), have been associated with HSAN-III [21]. This protein is required for RNA polymerase II complex assembly, and gene knockdown has been reported to inhibit peripheral neurogenesis, neuronal differentiation and development, and cerebral oligodendrocyte dyelination [22]. Homozygous IKBKAP knockout mice die in utero with abnormalities noted in both vascular and neural development [23].
Hereditary Sensory and Autonomic Neuropathy: Case Report and Discussion
Eric J. Wright1, Rebecca M. Garza1 and Derrick C. Wan1,2*
- 1Division of Plastic and Reconstructive Surgery, Department of Surgery, Stanford University School of Medicine, Stanford, CA, USA
- 2Hagey Laboratory for Pediatric Regenerative Medicine; Stanford University School of Medicine, Stanford, CA, USA
*Address for Correspondence: Derrick C. Wan, MD, Assistant Professor, Division of Plastic and Reconstructive Surgery, Department of Surgery, Stanford University School of Medicine, 257 Campus Drive West, Stanford, CA, 94305-5148, USA, Tel: 650) 723-7073; Fax: (650) 736-1705; E-mail: dwan@stanford.edu
Citation: Wright EJ, Garza RM, Wan DC. Hereditary Sensory and Autonomic Neuropathy: Case Report and Discussion. J Clin Med Case Reports. 2013;1(1): 4.
Copyright © 2013 Derrick C. Wan. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use,distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical & Medical Case Reports | ISSN 2332-4120 | Volume: 1, Issue: 1
Submission: 08 April 2013| Accepted: 18| June 2013| Published: 24 Jun 2013
Abstract
Hereditary sensory and autonomic neuropathies (HSAN) are a diverse group of diseases involving the peripheral nervous system. Patients present with profound distal sensory loss and variable degrees of autonomic disturbances. Multiple subtypes have been defined based on clinical symptoms and genetic testing. We present a case report of a nine month-old female with recurrent hand lesions and an absent response to noxious stimuli who was ultimately diagnosed with HSAN. A review of the literature and discussion of the pathogenetic mechanisms, current treatment, and future therapies for HSAN are also provided.Keywords
Neuropathy; Anhidrosis; Congenital insensitivity to pain; Sensory lossIntroduction
Hereditary Sensory and Autonomic Neuropathies (HSAN) comprise a heterogeneous group of disorders involving the peripheral nervous system. The first description of these conditions was made in 1852 with a report of three brothers with neurotrophic plantar ulcers [1]. In 1975, the term, "hereditary sensory and autonomic neuropathy," was introduced to reflect the substantial degree of autonomic involvement in these disorders [2]. Significant clinical variability exists among patients with HSAN, and while some present with pure sensory involvement, others demonstrate mixed sensory and motor involvement, minor autonomic disturbances, or pure autonomic neuropathies [3]. Most commonly, patients with HSAN present with loss of pain and temperature sensation which may lead to chronic ulcerations in the hands and feet. These wounds can progress to severe soft tissue infection or may lead to osteomyelitis with ultimate amputation of the affected limb.HSANs can be broadly subdivided based on mode of inheritance into autosomal dominant forms, with either juvenile or adult-onset, and autosomal recessive forms with congenital or early childhood onset [4]. More recent genetic testing has now allowed for subdivision into five types based on age at onset, mode of inheritance, predominant features, and genetic mutation [4]. In this case report, we present a patient with bilateral hand lesions, cellulitis, and tongue and lip lesions who was ultimately diagnosed with HSAN. A review of this rare condition and overview of insights gained from molecular and genetic studies are also discussed, with an emphasis made on the importance of proper diagnosis, need for family counseling, and potential future therapeutic interventions.
Case Report
A nine month-old female was brought in to the Emergency Department for evaluation of rapidly progressive skin lesions on the hands and a small resolving vesicular lesion on the tip of the tongue. The patient’s mother reported first noticing a small, red bump on the left thumb that grew and turned black over the course of one day. Small blisters were also noted on other fingers. Aside from these lesions, the patient’s mother reported no other symptoms. The patient was born full-term and had met gross motor milestones. Physical examination revealed a 2 x 1 cm black, well-demarcated eschar on the volar surface of the left thumb (Figure 1A). Other small 5 mm clear bullae were appreciated on the volar surface of left long finger. Yellow discharge and a surrounding border of blanching erythema were also observed. Interestingly, no tenderness to palpation was appreciated on manipulation of lesions. The remainder of the examination was consistent with normal motor function, as the child was able to sit and crawl on her own and hold small objects in her hand. In addition, the patient had 1+ patellar reflexes, downgoing toes bilaterally, and no clonus.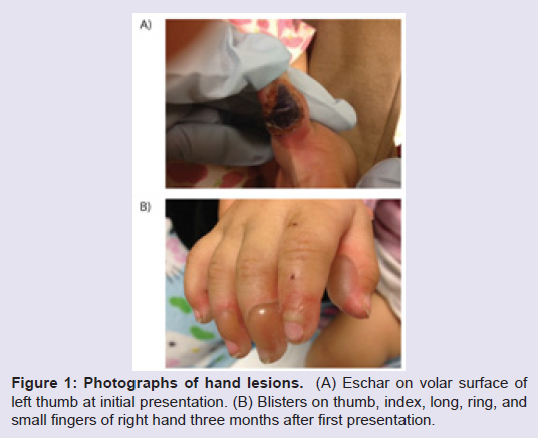
Figure 1: Photographs of hand lesions. (A) Eschar on volar surface of left thumb at initial presentation. (B) Blisters on thumb, index, long, ring, and small fingers of right hand three months after first presentation.
Three months later, the patient was brought back to the Emergency Department with similar development of multiple large blisters on the right hand (Figure 1B ). On further questioning, the patient’s mother reported the patient having a fever earlier in the day to 38.9° C but did not appreciate any sweating. In addition, despite multiple lesions on her right hand, the patient was noted to continue crawling in her usual fashion. She cried during physical examination, but crying was not specifically associated with inspection, palpation,or application of pressure to the hand lesions. Given these findings, along with prior negative workup, a neurology consult was obtained, and the patient was diagnosed with HSAN. The blisters were unroofed, and local wound care resulted in ultimate improvement of these lesions (Figure 2). In discussion with the patient’s mother, she reported no other family members with similar symptoms. She was instructed to provide close supervision of the patient and daily inspection of skin and mucocutaneous surfaces. The mother was also instructed to bring the patient back for medical attention should any new lesions develop.
Discussion
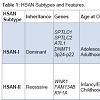
HSAN is an extremely rare clinical entity that is frequently missed due to lack of uniformity in presentation and lack of simple diagnostic measures. Pathogenesis involves progressive degeneration of sensory and autonomic neurons, resulting in ulcerative wounds and a variable degree of autonomic and/or motor disturbances. The genetic spectrum of HSANs include both autosomal dominant and recessive forms, and familial studies have identified 12 causative genes to date [3]. Some of these gene mutations have been causally linked. However, many remain poorly understood. Based on clinical presentation and genetic testing, five types of HSAN have been defined (Table 1).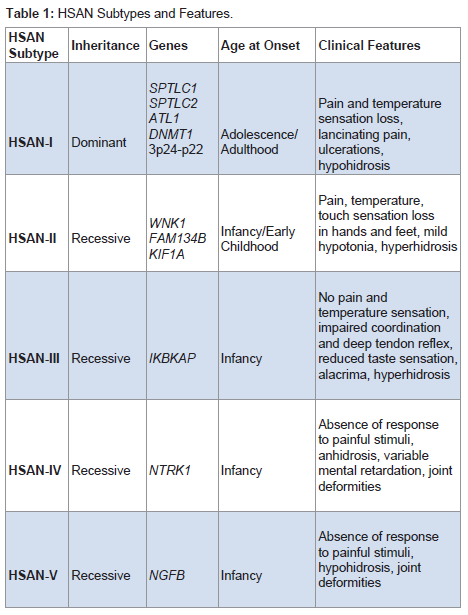
Table 1: HSAN Subtypes and Features.
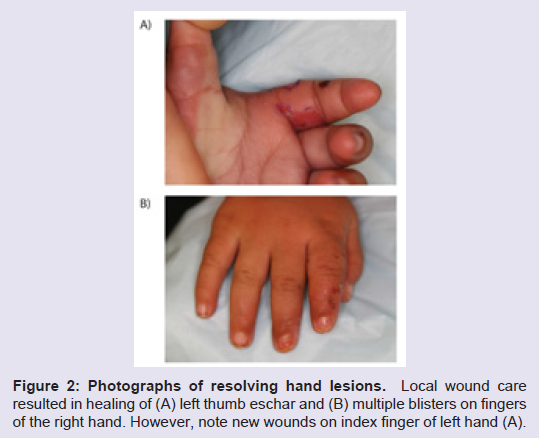
Figure 2: Photographs of resolving hand lesions. Local wound care resulted in healing of (A) left thumb eschar and (B) multiple blisters on fingers of the right hand. However, note new wounds on index finger of left hand (A).
HSAN-IV demonstrates an autosomal recessive inheritance pattern and is also referred to as congenital insensitivity to pain with anhidrosis (CIPA). Patients with HSAN-IV present with abnormal or absent responses to typically painful stimuli, anhidrosis particularly notable during fevers, variable degrees of mental retardation, and ultimately have a reduced life expectancy [3]. While only NTRK1 has been linked to HSAN-IV, over 40 different missense, nonsense, frameshift, and splice-site mutations have been identified [24,25]. NTRK1 encodes the high-affinity nerve growth factor receptor Trk-A which is involved in neurotrophin signaling [24]. Neurite outgrowth and survival of sympathetic ganglia and nociceptive sensory neurons have all been shown to be dependent on Trk-A signaling [24].
Lastly, patients with HSAN-V closely resemble those with HSAN-IV. Also demonstrating autosomal recessive inheritance, HSAN-V presents with absent responses to painful stimuli, and patients may have abnormal joints. However, unlike HSAN-IV, HSAN-V is associated with hypohydrosis, not anhidrosis, and no mental retardation [3]. Mutations in NGFB, which encodes β-NGF have been associated with HSAN-V 26]. Interestingly, β-NGF servesas a ligand for Trk-A, thereby explaining many of the similarities in presentation to HSAN-IV [27]. However, β-NGF has also been shown to signal through alternative receptors such as p75NTR, and altered activity through these other pathways may result in some of the more subtle differences observed between the two types [27,28].
In this patient, onset of sensory and autonomic neuropathies occurred in early infancy, and there were no other family members with similar symptoms. The patient likely inherited an autosomal recessive form of HSAN. Physical examination revealed normal motor tone and deep tendon reflexes. While the patient may be classified into HSAN-IV or HSAN-V, observations of anhidrosis during fevers and absent tears with crying are more congruent with a HSAN-IV presentation. Formal genetic studies remain to be performed, and both NTRK1 and NGFB mutations will need to be tested to definitively classify our patient, but the family has yet to consent to such testing. Such identification will be important not only to enable accurate diagnosis, but also to provide prenatal counseling for the mother and to guide genetic testing for subsequent pregnancies [29]. Currently, treatment of HSAN remains mainly supportive. Patients frequently suffer from recurrent ulcerations which may lead to osteomyelitis and eventual spontaneous or surgical amputations [3]. In the future, with increasing knowledge regarding disease pathogenesis, gene-specific therapies may be developed to target known protein deficiencies. Furthermore, with the advent of human-induced pluripotent stem cells, new models are being actively developed to assist in disease modeling and drug screening for the treatment of HSANs [30].
References
- Nelaton A (1852) Affection singuliere des os du pied [French]. Gazette Hopitaux Civils Militaires (Paris) 4: 13.
- Dyck PJ, Thomas PK, Griffin JW, et al., eds. (2005) Peripheral Neuropathy. Philadelphia: WB Saunders.
- Rotthier A, Baets J, Timmerman V, Janssens K (2012) Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol8: 73-85.
- Khaledi M, Rezaei N (2012) Hereditary and sensory autonomic neuropathies. Iran J Pediatr 22: 567-568.
- Rotthier A, Baets J, De Vriendt E, Jacobs A, Auer-Grumbach M, et al. (2009) Genes for hereditary sensory and autonomic neuropathies: a genotypephenotype correlation. Brain 132: 2699-2711.
- Auer-Grumbach M (2008) Hereditary sensory neuropathy type I. Orphanet J Rare Dis 3: 7.
- Hanada K (2003) Serine palmitoyl transferase, a key enzyme of sphingolipid metabolism. Biochim Biophys Acta 1632: 16-30.
- Tsui-Pierchala BA, Encinas M, Milbrandt J, Johnson EM Jr (2002) Lipid rafts in neuronal signaling and function. Trends Neurosci 25: 412-417.
- Penno A, Reilly MM, Houlden H, Laurá M, Rentsch K, et al. (2010) Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285: 11178-11187.
- Guelly C, Zhu PP, Leonardis L, Papić L, Zidar J, et al. (2011) Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am J Hum Genet 88: 99-105.
- Zhu PP, Soderblom C, Tao-Cheng JH, Stadler J, Blackstone C (2006) SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum Mol Genet 15: 1343-1353.
- Durr A, Camuzat A, Colin E, Tallaksen C, Hannequin D, et al. (2004) Atlastin1 mutations are frequent in young-onset autosomal dominant spastic paraplegia. Arch Neurol 61: 1867-1872.
- Klein CJ, Botuyan MV, Wu Y, Ward CJ, Nicholson GA, et al. (2011) Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet 43: 595-600.
- Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28: 1057-1068.
- Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM (2012) Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry 83: 119-120.
- Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, et al. (2009) Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet 41: 1179-1181.
- Lafreniere RG, MacDonald ML, Dube MP, MacFarlane J, O’Driscoll M, et al. (2004) Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am J Hum Genet 74: 1064-1073.
- Fu Y, Subramanya A, Rozansky D, Cohen DM (2006) WNK kinases influence TRPV4 channel function and localization. Am J Physiol Renal Physiol 290: F1305-F1314.
- Riviere JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, et al. (2011) KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 89: 219-230.
- Axelrod FB (2004) Familial dysautonomia. Muscle Nerve 29: 352-363.
- Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, et al. (2001) Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 68: 598-605.
- Otero G, Fellows J, Li Y, de Bizemont T, Dirac AM, et al. (1999) Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell 3: 109-118.
- Chen YT, Hims MM, Shetty RS, Mull J, Liu L, et al. (2009) Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol Cell Biol 29: 736-744.
- Indo Y (2002) Genetics of congenital insensitivity to pain with anhidrosis (CIPA) or hereditary sensory and autonomic neuropathy type IV. Clinical, biological and molecular aspects of mutations in TRKA(NTRK1) gene encoding the receptor tyrosine kinase for nerve growth factor. Clin Auton Res 1: I20-I32.
- Sarasola E, Rodriguez JA, Garrote E, Arístegui J, García-Barcina MJ (2011) A short in-frame deletion in NTRK1 tyrosine kinase domain caused by a novel splice site mutation in a patient with congenital insensitivity to pain with anhidrosis. BMC Med Genet 12: 86.
- Einarsdottir E, Carlsson A, Minde J, Toolanen G, Svensson O, et al. (2004) A mutation in the nerve growth factor beta gene (NGFB) causes loss of pain perception. Hum Mol Genet 13: 799-805.
- Carvalho OP, Thornton GK, Hertecant J, Houlden H, Nicholas AK, et al. (2011) A novel NGF mutation clarifies the molecular mechanism and extends the phenotypic spectrum of the HSAN5 neuropathy. J Med Genet 48: 131- 135.
- Covaceuszach S, Capsoni S, Marinelli S, Pavone F, Ceci M, et al. (2010) In vitro receptor binding properties of a “painless” NGF mutein, linked to hereditary sensory autonomic neuropathy type V. Biochem Biophys Res Commun 391: 824-829.
- Couzin-Frankel J (2010) Chasing a disease to the vanishing point. Science 328: 298-300.
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, et al. (2009) Modelling pathogenesis and treatment of familial dysautonomia using patientspecific iPSCs. Nature 461: 402-406.