
Journal of Metabolomics & Systems Biology
Download PDF
Review Article
*Address for Correspondence: Troy D. Wood, Department of Chemistry, University at Buffalo, State University of New York, Buffalo, NY14260-3000, Tel: (716) 645-4144; Fax: (716) 645-6963; E-mail: twood@buffalo.edu
Citation: Rudolph HL, Friesen WL, Wood TD. The Hunt for Biomarkers of Autism Spectrum Disorders. J Metabol Sys Biol. 2013;1(1): 11.
Copyright © 2013 Troy D. Wood et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 1, Issue: 1
Submission: : 31 May 2013 | Accepted: 03 July 2013 | Published: 06 July 2013
De novo mutations tend to be paternal in origin and correlate with the father’s age [51,52]. O’Roak et al. sequenced the coding regions of the genome for parent-child trios and some of the unaffected siblings,and showed that de novo point mutations are overwhelmingly paternal (4:1) in origin for ASD [51]. The paternal origin of single point mutations and small indels in ASD is also supported by the results of Iossifov et al., who additionally show that frequency of de novo mutation is associated with a parent’s age, with older parents having more de novo mutations [52]. Indeed, a report by Kong et al. indicates that diversity in mutation rate of single nucleotide polymorphisms is dominated by the age of the father at conception of the child and hence implicated as a major factor in development of ASD in offspring [53].

In another protein analysis study, hair and nail samples from ASD and controls were analyzed by SDS-page and Western blot to assimilate a correlation between sulfur-containing proteins and the percent of nitration of keratin proteins [71]. From this study, it was demonstrated that sulfur-containing proteins in hair were decreased and nitrogen-containing proteins were increased in ASD compared to controls. With regard to sulfur-containing proteins, it is an interesting correlation to oxidative stress of GSH in ASD individuals (discussed below). The decrease in sulfur-containing proteins in hair and nails in ASD could be due to dysfunction in the regulation of sulfurcontaining amino acids, such as that shown in the γ-glutamyl cycle (Figure 3). The increase in nitrogen-content in proteins with regard to ASD could also be correlated to the increase of NO species due to oxidative stress (also discussed below). In addition to these findings, there is a correlation with regard to the severity of ASD and the concentration of nitrogen-containing proteins and sulfur-containing proteins. With regard to sulfur-containing proteins, individuals with severe forms of ASD (LFA) had the lowest concentration of sulfurcontaining proteins compared to less severe forms of ASD (HFA) which had a relatively higher concentration. The opposite was shown with regard to nitrogen-containing proteins where LFA individuals had the higher concentration and HFA had a lower concentration.
Another potential protein of interest is the high mobility box group 1 (HMBG1) protein, a ubiquitous protein that can act as an activator for the immune response or signal for induced inflammation [72]. More importantly, it has also been shown that an increase of [HMBG1] in the cytosol may be due to glutamate excitotoxicity, which correlates with the oxidative stress theory previously discussed [73]. With these correlations, Emanuele et al. have shown that there is an increase in [HMBG1] with ASD [74]. They have also found a potential correlation with regard to the higher [HMBG1] protein with respect to the severity of ASD phenotype symptoms.

A final study that is discussed here is based on the work performed by Corbett et al. that uses liquid chromatography coupled to electrospray ionization mass spectrometry (LC-ESI-MS) for identification of possible proteins as biomarkers in ASD [79]. From their work, four potential protein biomarkers were found: apolipoprotein (apo) B-100, complement factor H related protein (FHR1), Complement C1q, and Fibronectin 1 (FN1). These proteins serve a variety of different functions including but not limited to lipid transportation, phagocytosis and cell mediation. The results from this study are discussed in Table 1 and demonstrate a negative correlation of ASD to the [apo B-100] and a positive correlation of ASD with respect to [FHR1], [Complement C1q] and [FN1]. This study also demonstrated that concentrations of the studied proteins are closer to controls with HFA subjects compared to LFA subjects.

*Values reported in this paper were as GSH and GSSG and not the ratio of GSH:GSSG. The following is the actual reported values:

Nitrogen oxide

Lipid oxidative stress

The curious case of Stercobilin

aStatistically different from severely autistic group (p<0.00000003).
The Hunt for Biomarkers of Autism Spectrum Disorders
Heather L. Rudolph, William L. Friesen, and Troy D. Wood*
- Department of Chemistry, University at Buffalo, State University of New York, Buffalo, NY 14260-3000, USA
*Address for Correspondence: Troy D. Wood, Department of Chemistry, University at Buffalo, State University of New York, Buffalo, NY14260-3000, Tel: (716) 645-4144; Fax: (716) 645-6963; E-mail: twood@buffalo.edu
Citation: Rudolph HL, Friesen WL, Wood TD. The Hunt for Biomarkers of Autism Spectrum Disorders. J Metabol Sys Biol. 2013;1(1): 11.
Copyright © 2013 Troy D. Wood et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Metabolomics & Systems Biology | ISSN: 2329-1583 | Volume: 1, Issue: 1
Submission: : 31 May 2013 | Accepted: 03 July 2013 | Published: 06 July 2013
Abstract
Autism spectrum disorders (ASD) are a group of neurodevelopmental disorders in which individuals may experience delayed or impaired language development as well as difficulties in social interactions. Diagnosis is made on the basis of a series of psychological tests and observed behavior; a clinical biochemical diagnosis does not yet exist for ASD. Advances in modern technologies in the fields of genomics, proteomics, and metabolomics are adding to our knowledge of ASD etiology. This review examines the progress in identifying potential biomarkers of ASD, particularly in the past decade; the advancements made in the areas of genomics, proteomics, and metabolomics indicate that ASD are inherently heterogeneous, yet there may yet be optimism that in spite of this heterogeneity, one or several markers may emerge to advance clinical diagnosis and eventually treatment of ASD.Keywords
Autism; ASD; Biomarker; Genomics; Proteomics; Metabolomics; Oxidative stress; Gene arrays; Mass spectrometryIntroduction
Autism spectrum disorders (ASD) [1] are characterized by impaired social interactions, deficits in communications skills, repetitive behaviors, and other stereotypical behavioral patterns [1-6]. ASD are presumed to be etiologically and biologically heterogeneous [7]. In early onset autism (EOA), also known as “infantile” ASD, symptoms begin in infancy [2,8]; this type of ASD, first reported by Dr. Leo Kanner [1,2], has sometimes been referred to as “classical” ASD. With a second subtype dubbed “regressive” ASD, development appears to be normal until regression into ASD symptoms, typically between 18-36 months of age [9-11]. However, it should be noted that often there are some indications of minimal ASD symptoms being present in these individuals before the onset of “regressive” behaviors, based on video observations of old home movies [12,13].ASD diagnosis is made through behavioral observation and a battery of psychological tests based on the criteria for autistic disorder as defined in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV), and thus has a subjective element [14-16]. Diagnosis is often very difficult for children that are under the age of two, and can be complicated by the existence of additional medical problems [17,18]. ASD have become diagnosed far more frequently in recent years than in previous decades [19-22]. Recent data from the National Survey of Children’s Health shows that the incidence of ASD in the United States is now 1 in 50 in 2011-12 [23]. Now, whether this is due to a true increase in the incidence of ASD, due to broadening of ASD diagnostic criteria, or increases in awareness and understanding of the diagnostic criteria for ASD, remains an open question [19]. In either case, ASD are common in the United States and are therefore a serious public medical concern. Behavioral intervention becomes increasingly effective for persons with ASD the earlier it is introduced [24-27], and consequently approaches that can accurately diagnose ASD at earlier ages are of utmost importance. The lifetime cost of ASD per capita has been estimated at $3.2 million [28].
It has long been a goal to discover and validate potential biomarkers for ASD. Identification of biomarkers would not onlyprovide insight into the underlying mechanisms responsible for the development of ASD, but it would also generate potential therapeutic targets for treatment of ASD and provide additional objective diagnostic criteria for ASD, which would accelerate the introduction of behavioral therapy. The purpose of this review is to examine progress toward identifying ASD biomarkers. Naturally, such a review will examine potential ASD causative factors. While a single cause of ASD has not been identified, research has emerged that implicates genetic, epigenetic, and environmental contributions to neurodevelopment that either cause (or may confer) susceptibility to developing ASD [29,30].
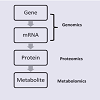
Our discussion will follow the “omics cascade” shown in Figure1 [31], starting with markers at the genomic level, followed by proteomic markers and metabolomic markers. Of course, any of the biological factors can additionally be influenced by environmental factors. In particular, several metals have emerged which may have clinically-relevant diagnostic value and a role in ASD etiology and will also be discussed.
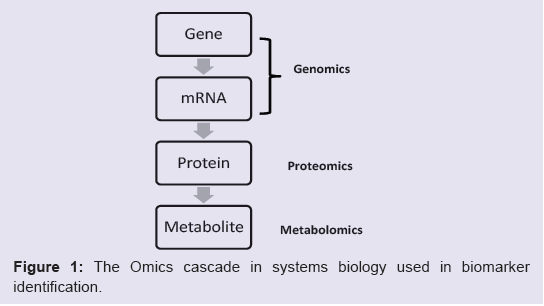
Figure 1: The Omics cascade in systems biology used in biomarker identification.
Genetic Basis of ASD
Because of the heterogeneity of ASD, it has remained a challenge of modern medicine to decipher its etiology. In medicine, when causation of a disorder is unknown, twin and family studies are used to establish whether any genetic risk factors may be involved. Studies of monozygotic twins have revealed a concordance rate of 36-96% [32-38], while dizygotic twins showed a concordance rate of 0-30% [32-38]. Thus, heritability of ASD has been estimated to be as high as >85% [39], though others posit a more conservative estimate of heritability slightly below 40% [38]. In any case, the less than 100% concordance of monozygotic twins suggests genetic factors alone do not dictate the phenotypic expression of ASD, and that environmental factors at least weakly influence the phenotype expressed.Sibling studies of ASD incidence are useful for two reasons. First, they can have larger subject cohorts, and second, they can be compared to the studies of dizygotic twins. From 99 ASD probands, a 2.9% concordance amongst siblings has been reported [40]. A much larger study, including over 2900 children, by Constantino et al. indicated that 10.9% of families with a child diagnosed with ASD had at least one more child with ASD, and that 8.2% of all siblings of ASD children were also symptomatic [41]. A more recent study from Constantino et al. indicates that half-siblings show about one-half the incidence rate as full siblings [42]. Overall, the recurrence risk to siblings is 5-10x that of the general population [43-45]. DNA analysis of individuals with shared ancestry has also implicated individual gene loci in ASD [46]. Thus, twin and other family studies provide strong evidence of a genomic component to ASD etiology.
ASD Genomics
As noted by Geschwind, the past decade “has brought an explosion of genetic findings in ASD” [47]. As genomics methods emerged to detect single nucleotide polymorphisms (SNPs), there was some initial optimism that by collecting DNA from families containing one or more persons with ASD that genomics would rapidly indicate potential genes that might predispose individuals to developing ASD. Part of this optimism was because it had been well established that single chromosome abnormalities including Fragile X syndrome, Tuberous Sclerosis, Down’s syndrome, Smith-Lemli-Optiz syndrome, Timothy syndrome, phenyl ketonuria and dozens of others are associated with autism [48]. All of these are rare genetic variations and in total account for perhaps as much as 10% of ASD incidence.In fact, genomics research has indicated that underlying genetic causes of ASD are highly complex [49]. Figure 2 is a pie chart representing the relative distribution of ASD causes. Miles notes that a genetic cause can only be identified in 20-25% of ASD cases; save for a small percentage of teratogenic exposures, the cause of the remaining 75-80% is unknown [50]. Thus, Mendelian (single-gene) mutations cannot account for the vast majority of instances of ASD, although they do account for approximately 5% of total incidence [50]. Another 5% of incidence is due to chromosomal abnormalities which are cytogenetically visible [50]. These single gene/single chromosome disorders represent an array of different underlying molecular processes and mechanisms. Regardless, each is relatively uncommon. This has led to speculation that autism may simply be a broad descriptor of a much larger set of conditions possessing different etiologies. One might even argue that if this is the case, that searching for an ASD genotype will be extraordinarily difficult.

Nevertheless, the evidence continues to mount that insertions and deletions in the genome known as copy number variants (CNVs) account for a significant fraction of ASD incidence [54], accounting for between 10 and 20% [50]. CNVs are often inconsequential because the regions duplicated or deleted contain no genes, or because of a functional compensation for the quantity of the expressed gene; however, those CNVs which occur in functionally important regions can lead to disease. A growing database containing CNVs has been in progress for the past few years [54] and can be accessed at http:// projects.tcag.ca.org. Known structural variants associated with ASD exist on every chromosome. While some of these CNVs are inherited, others are de novo and are highly associated with ASD [55,56]. Thus, it could be argued that, ASD are heritable, but not inherited [57].
In a large scale study, the Autism Genome Project (AGP) analyzed 1000 subjects with ASD and 1300 controls, and found that individuals with ASD are much more likely to contain CNVs than their counterparts [58]. One of the first CNVs associated with ASD was the microdeletion on chromosome 16p11.2 [59]. De novo CNVs associated with ASD implicate genes involved in the formation and function of synapses [60] and neurons [61,62]. Rare CNVs have been implicated for ASD not only at 16p11.2, but at 7q11.23, 15q11.2-13.1, and Neurexin 1[63].
Males develop ASD almost at four times the rate of females [64]. The reason for this imbalance is unclear, and it is tempting to attribute this to genetics, although a gender-linked biomarker has not yet been established. A recent European study suggests a female protective effect from developing ASD [65].
Dizygotic twin pairs were examined, and siblings of females with traits for autism showed more traits for autism than siblings of males, which suggests females may require greater familial etiologic load to manifest phenotypical expression of ASD [65].With ASD exhibiting such genomic heterogeneity, it may be impossible to define a single or limited number of genomic markers with which to characterize and diagnose it. In spite of this, persons with ASD share phenotypic traits. Therefore, is it possible that inspite of the genetic heterogeneity there are underlying molecular mechanisms that account for the manifestation of ASD. Inborn errors of metabolism account for less than 5% of all cases of ASD [66]. This provides the tantalizing prospect that the emerging field of metabolomics may yet play a role in defining potential ASD biomarkers, which will be discussed later in this review.
Proteins Associated with ASD
Proteomic research has proven to be a powerful tool to correlate disease detection with protein abnormalities. Even with the power of genomics, proteomics can provide additional value to clinical diagnostics because proteins translated from the genome can undergo post-translational modifications, formation of disulfide bonds, and proteolytic processing that may provide indications of disease. Potential protein biomarkers have been studied for a number of disease states including Alzheimer’s, cancer, and diabetes, to name a few [67-69]. ASD has also been associated with protein research for the relation of potential biomarkers (Table 1). One study has found a potential correlation between the protein secreted amyloid precursor protein-α (sAPPα) and individuals who are afflicted with severe forms of ASD [70]. In this study, severe ASD individuals were compared to controls for sAPPα concentration. Using ELISA and Western blot techniques, blood samples were analyzed and it was shown that sAPPα levels are higher in individuals with ASD as compared to the controls. Another unique aspect of this study is that they began to attempt to use their findings as a means for early detection of ASD. Using blood samples from 150 umbilical cords, they performed the same analysis to determine the concentration of sAPPα within these samples as a means for a potential early detection of ASD. In this study, 7% of the umbilical cords exhibited an increased concentration of sAPPα. A follow up study with regard to the 7% increase of this protein is warranted to validate sAPPα as a potential biomarker for early detection of ASD.Table 1: Potential protein biomarkers for ASD diagnosis.
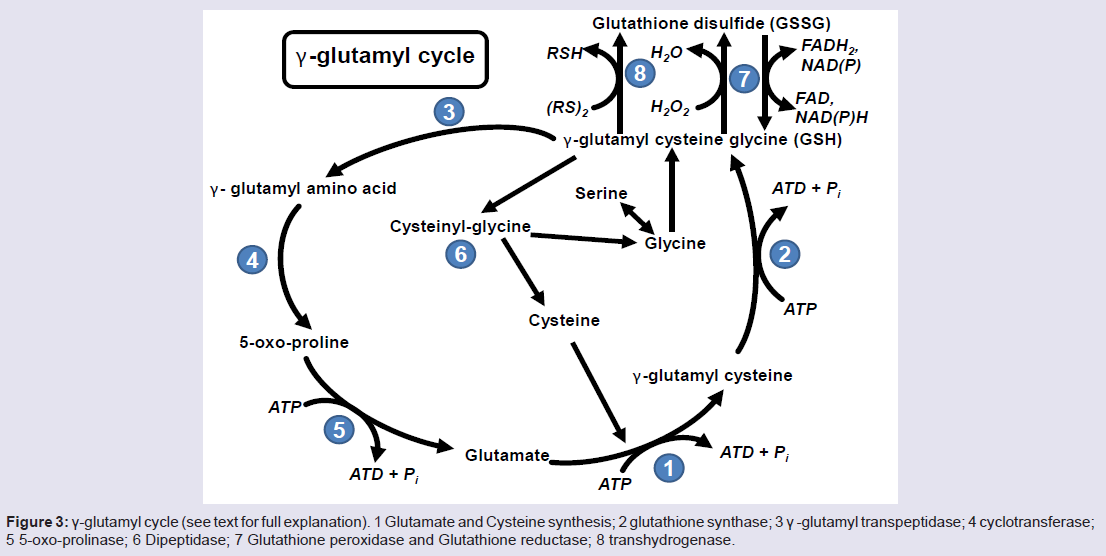
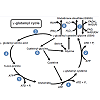
Figure 3: γ-glutamyl cycle (see text for full explanation). 1 Glutamate and Cysteine synthesis; 2 glutathione synthase; 3 γ -glutamyl transpeptidase; 4 cyclotransferase; 5 5-oxo-prolinase; 6 Dipeptidase; 7 Glutathione peroxidase and Glutathione reductase; 8 transhydrogenase.
One protein that has received considerable recent attention as an ASD biomarker is engrailed-2 (EN2). EN2 is a homeobox transcription factor located on chromosome 7q36 that is important in the development of the central nervous system (CNS). Several studies have reported that transgenic mice in which EN2 has been overexpressed show a decrease in the number Purkinje cells, a smaller cerebellum, and hypoplasia [75-78]. Two intronic EN2 single nucleotide polymorphism (SNP) have been found to be associated with ASD (rs1861972 and rs1861973). These SNP alleles have found to be over expressed in ASD individuals and under representative in their paired siblings [77]. In addition to sibling genetic comparisons, it has also been shown that these alleles are overexpressed in ASD individuals compared to controls [76,78]. Table 2 is a brief summary of the relationship of alleles on EN2 and their correlation to ASD.
Table 2: EN2 SNP correlation to ASD individuals.
Metabolic Indicators of ASD
Because the underlying genomics of ASD is heterogeneous while there are expressed similarities in ASD symptoms makes it natural to suspect that the genomic influence may be expressed as a metabolic imbalance. In the past decade, significant progress has been made toward developing phenotypic metabolic panels for ASD. Much of this research has revealed systemic oxidative stress in individuals with ASD. This will now be discussed.
Oxidative stress
Several studies have been performed studying the role of oxidative stress on cells and tissues with respect to ASD. Oxidative stress is the process by which reactive oxygen species (ROS) oxidize key components of cells and tissues. These reactions include, but are not limited to, oxidation of polyunsaturated fatty acids, molecular species containing thiols, and in some cases the reduction of Ferriccontaining proteins and nitric oxide [80]. Several of these oxidative species and processes have been examined with respect to the onset of ASD.
Glutathione
Glutathione (GSH) is a tripeptide consisting of L-γ-gluamyl-Lcysteinyl- glycine with a mol. wt. of 307 g/mol [81]. Glutathione acts as an antioxidant in cell systems, helping regulate the oxidized form of GSH, glutathione disulfide (GSSG) or a glutathione-S-conjugate (Figure 3). The first reaction involved with the formation of GSH is the ATP dependent reaction between glutamate and cysteine to form γ-glutamyl cysteine (Figure 3, Reaction 1) which occurs intracellularly. This γ-glutamyl cysteine then undergoes another ATP dependent reaction where glycine binds to γ-glutamyl cysteine via glutathione synthase to form GSH (Figure 3, Reaction 2). GSH can either be oxidized or it can undergo degradation via γ -glutamyl transpeptidase where it is exported out of the cell and cleaves to form cysteine-glycine and γ-glutamyl amino acid moiety (Figure 3, Reaction 3). The γ-glutamyl amino acid moiety can then travel back into the cell via cyclotransferase which cleaves the amino acid to form 5-oxo-proline (Figure 3, Reaction 4). ATP then catalyzes the reaction of 5-oxo-proline to form glutamate with 5-oxo-prolinase (Figure 3, Reaction 5), which is the starting precursor for the γ-glutamyl cycle [82,83]. The cysteine-glycine that was cleaved during the degradation of GSH (Figure 3, Reaction 3) undergoes further degradation into cysteine and glycine by dipeptidase (Figure 3, Reaction 6). Once GSH is formed, it can undergo oxidative stress via two different pathways, either with transhydrogenation (Figure 3, Reaction 8) or by selenium containing GSH peroxidase (GSH-PX) (Figure 3, Reaction 7). During oxidative stress, GSH is oxidized to GSSG via GSH-Px which catalyzes the reduction of H2O2 into H2O [83]. GSSG can then be reduced to GSH by glutathione reductase (GSH-R) with NAD(P)H co-factor (Figure 3, Reaction 7) [80,82,83].
Normal cells have a higher ratio of GSH: GSSG, indicating there to be little to no oxidative stress. Autism studies have overwhelmingly shown that the balance of GSH: GSSG is reduced compared to their normal cell studies [82,84-87]. Studies have also shown that the concentration of GSH-Px in ASD specimens to be lower than that of controls, indicating that it has been used to oxidize GSH, and that there is oxidative stress occurring [88,89]. The methods to quantify the GSH: GSSG to be lower in individuals with ASD has varied from spectrometry techniques to spectrophotometric techniques. The sample types that have demonstrated these results have also varied from tissue, blood, and urine specimens [87-94]. Within these select studies that were reviewed, our focus was on the comparison on the ratio of GSH: GSSG between diagnosed ASD individuals and their paired controls (Table 3). Within these studies, ASD subjects were either compared to age/gender matched, non-ASD diagnosed individual [87,90-92,94], or a non-ASD sibling [93]. In both situations, ASD subjects showed a 30-50% decrease of GSH: GSSG compared to the non-ADS individuals. It is also prevalent that all methodologies and sample types used to quantify the GSH: GSSG demonstrate the same trend that ASD individuals have a marked decrease of GSH: GSSG.While each individual study had a small sample set of ASD and non-ASD individuals, the combined results begin to show a prevalent correlation between GSH oxidative stress and ASD diagnosis.
Table 3: GSH: GSSG comparison in ASD vs. Control, values obtained by different methods and samples.
a. GSH: 3.23 ± 0.48 nmol/mL and GSSG: 0.447 ± 0.13 nmol/
b. GSH: 4.09 ± 0.79 nmol/mL and GSSG: 0.362 ± 0.10 nmol/mL
In addition to examining studies quantifying GSH: GSSG, we also reviewed studies measuring the concentration of GSH-Px (Table 4). As mentioned earlier, GSH-Px catalyzes the reaction that oxidizes GSH to GSSG. If oxidative stress occurs with relation to ASD, it would be expected that the overall concentration of GSH-Px would be decreased due to the fact that it is being used to oxidize GSH. From the results shown in Table 4, the overall [GSH-Px] in children with ASD is ~25% less than that of a non-ASD child. One study performed by Sogut et al. has shown a difference in this trend, that the [GSH-Px] is lower than that of the control group [95]. After comparing their methodologies with other studies that are reported, one difference is that blood is collected in a heparin-containing vial [95] as opposed to EDTA. The other difference is that Sogut et al. only used plasma to determine GSH-Px concentrations [95] where as other groups used both plasma and red blood cells for their analyses [71,88,89]. The correlating studies also measured GSH-Px using Ellman’s reagent or another type of spectrophotometric assay [71,88,89], whereas Sogut et al. determined GSH-Px by the addition of H2O2 to a reaction mixture of GSH, NADPH and GSH-R [95]. While the majority of studies shows a correlation of GSH oxidative stress, and is comparable to the studies described above, the sample set is very small and further work needs to be done.
Table 4: [GSH-Px] comparison in ASD and non-ASD individuals.
Nitrogen oxide (or nitric oxide, NO) is a non-polar molecule produced from arginine by a Ca2+ dependent nitrogen oxide synthase (NOS) [96]. NO has many important functions including acting as a vasodilator, aiding in neurotransmission, and potentially playing a role within immune systems [97]. Although NO is vital for normal activity within the human system, there have been studies correlating an increase in [NO] in individuals who have been diagnosed with ASD [71,95,97,98]. Two proposed pathways have been suggested as to explain why [NO] is increased in ASD individuals. The first proposed mechanism is related with Gamma-amino butyric acid (GABA) and GABA transaminase (GABA-T). GABA is considered to be a major inhibitory neurotransmitter and has also been associated with other neurological diseases such as schizophrenia, depression, alcoholism, and seizure disorders [98]. A study performed by Cohen et al. has suggested that increased [NO] levels inhibit the function of GABA-T, which is responsible for the degradation of GABA. Cohen’s findings demonstrate that not only is there an increase in [NO], but there is also an increase in GABA in ASD cases compared to the controls [98-100].
The second proposed mechanism associating increased [NO] levels in ASD individuals is the relationship between NO and GSH. GSH function, as discussed previously, serves to regulate oxidative stress within biological systems. In order to control [NO] in biological systems and to prevent oxidative stress, GSH can bind to NO to form S-nitrosylglutathione (GSNO) [101]. GSNO can be metabolized by GSH dependent formaldehyde dehydrogenase to form GSSG, ammonia and water while simultaneously oxidizing NADH to NAD+. This excess ammonia production may attribute to the increased [NO] in ASD individuals, as well as correlate with the increase of GSSG in ASD biological systems [102]. Another study shows that mean nitrite (a metabolite of NO) and adrenomedullin levels are higher in plasma of individuals with ASD than controls, although there is no correlation between total nitrite and adrenomedullin levels [103].
As established in the literature, there is a positive correlation with respect to [NO] and ASD. Table 5, entitled [NOx] species comparisonin ASD individuals versus non-ASD individuals, compares the results from this literature review. All references used the Griess reaction followed by spectrophotometric techniques to measure [NO] in blood.
Table 5: [NOx] species comparison in ASD individuals versus non-ASD individuals.
Another potential relationship to ASD is the oxidative stress of lipids. Due to the hydrophobic nature of lipids, they are effective barriers toward polar molecules, in addition to possessing many other important properties [96]. Lipid oxidation is a multistep process in which lipids form lipid peroxide species [104]. This process can take place during oxidative stress, in which lipids react with free ROS to form lipid peroxides and hydrocarbon polymers. These lipid peroxide species can then be degraded by transition metals and metal complexes such as iron or hemoglobin. If the resulting lipid peroxides do not degrade rapidly, they can be harmful to cells due to the alteration of cell oxidation reduction balance [105].
The correlation between lipid oxidation and ASD has been an emerging area of research in recent years and has supplied some of the most promising ASD biomarker candidates. Meguid et al. have shown that malondialdehyde (MDA), which is a marker of lipid peroxidase, to be of a higher concentration in ASD individuals than the corresponding controls [88]. Chauhan et al. have also examined the effects of lipid oxidation with respect to ASD [105]. From their findings, the levels of MDA are higher in ASD individuals as compared to non ASD individuals. Another point of interest to note is that in this study the ASD individuals were the siblings of the control group, suggesting that this form of oxidative stress is correlated to the disease and is not expressed the same way in closely related family members. Al-Yafee et al. have correlating studies that support the relationship of lipid oxidative stress with ASD as well [87]. The difference between the aforementioned studies is that lipid oxidation was measured by Peroxiredoxin (Prx) I and III as opposed to MDA. Prx plays an important role in regulating cellular H2O2, redox signaling and apoptosis. While this potential biomarker is not exactly the same as the above mentioned in this section, it does follow the same trend that the [Prx] I and III are higher in ASD inflicted individuals in comparison to the controls. The studies which indicate lipid oxidation in ASD specimens are summarized in Table 6.
Table 6: Lipid oxidative stress studies with relation to ASD.
The majority of research with regard to the relationship of oxidative stress and ASD for the search for a biomarker has overwhelmingly focused on GSH, NO, and lipid peroxidase. It is also of interest that the majority of these studies have been analyzed using spectrophotometric techniques and with the use of blood samples. While these techniques are of importance and have a higher sensitivity than other analytical methodologies, it would be beneficial to pair these studies with spectrometric analysis to correlate structures of proteins, genes, and metabolites with the already studied spectrophotometric analyses.
With this in mind, research has been performed by our group using liquid chromatography-mass spectrometry (LC-MS) to detect candidate markers in biofluids. We were intrigued by the neurochemical theory put forth by Panksepp that ASD behaviors might be induced by exposure to exogenous substances from diet that mimic the action of endorphins, now known as the “opioid excess theory” [106]. Clinical indications that a subset of children with ASD had improved behavior upon embarking on the gluten-free caseinfree (GFCF) diet gave further credence to the opioid excess theory [107,108]. We began by looking for evidence of gluten exorphins [109] in urine and blood. After developing LC-MS methods for the detection of gluten exorphins in cerebrospinal fluid (CSF) [110,111], it became clear that gluten exorphin B5 degrades very rapidly in biological fluids [111]. In the presence of protease inhibitors, we could detect gluten exorphins B4 and B5 in human plasma for the first time [112]. However, the rapid degradation rates of exorphins in human plasma suggest that they are unlikely to be significant in the pathogenesis of most cases of ASD.
However, during the course of these investigations, while profiling urine specimens of ASD and control children for gluten exorphins, we came upon an intriguing discovery. As shown in Figure 4, LC-MS/ MS showed the presence of a substance in control urines at nominal m/z 595 that is abundant in control urine but depleted in ASD urines, on average to only 33% of controls [113]. Although it shares the same nominal mass as gluten exorphin B5, its MS/MS fragmentation pattern established it was another substance. Eventually, we identified it as stercobillin, an optically active metabolite that is formed by the hydrogenation of bilirubin during the process of heme breakdown [114,115]; final conversion to stercobilin is accomplished through the action of bacteria in the colon. Although the initial sample set was small (n = 7), it showed promise that [Stercobillin] may be useful as a putative metabolic biomarker of ASD, at least in a subset of individuals. Studies are continuing in our laboratories with larger opulations to validate whether urinary stercobilin may be a general ASD biomarker. Stercobilin depletion could be related to oxidative stress, either at the level of the bilirubin (an oxidatively sensitive molecule) [116], at the level of the bacteria colonizing the colon, or due to some other factor.
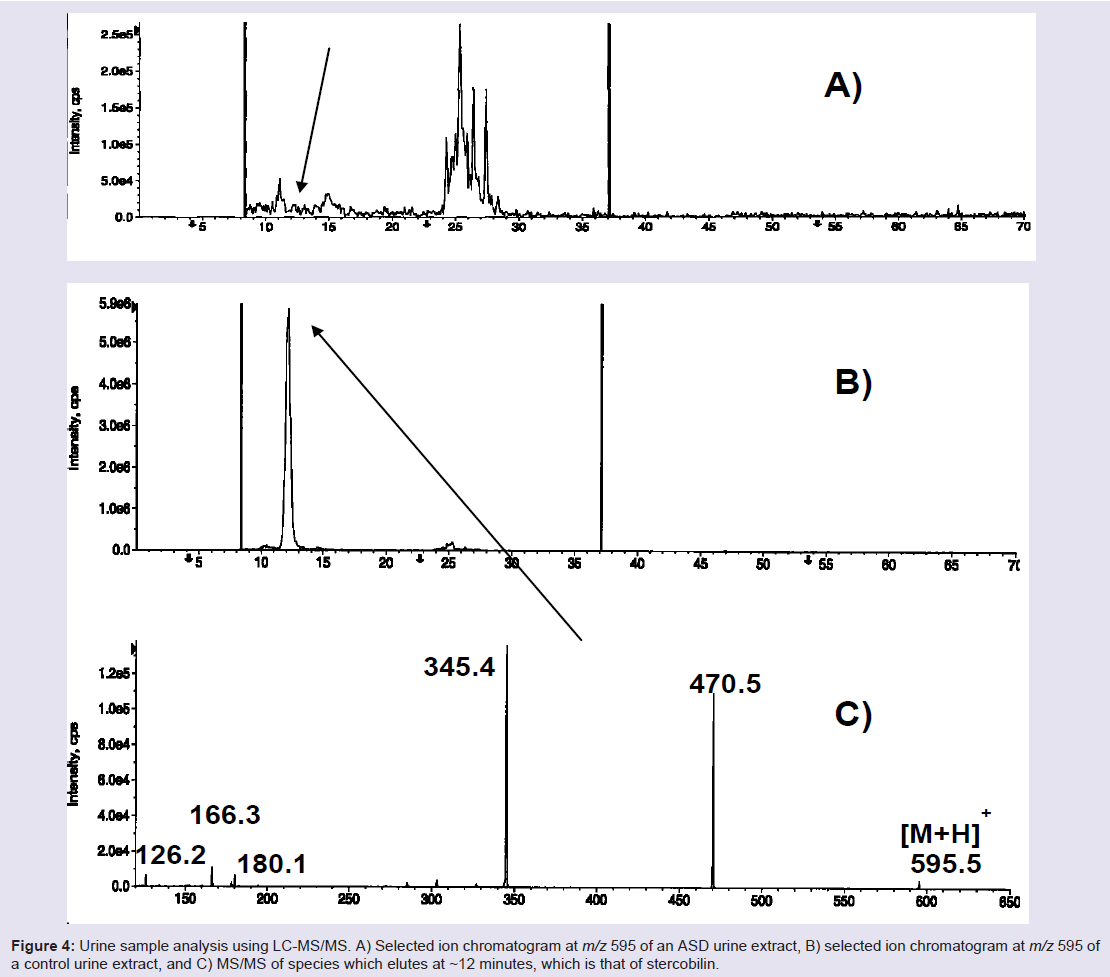
Figure 4: Urine sample analysis using LC-MS/MS. A) Selected ion chromatogram at m/z 595 of an ASD urine extract, B) selected ion chromatogram at m/z 595 of a control urine extract, and C) MS/MS of species which elutes at ~12 minutes, which is that of stercobilin.
Metals as Potential ASD Markers
While most of the species investigated as ASD markers fall under the Omics cascade, it is important to note that externally-derived species from environmental exposure are still likely to play some role in ASD etiology, as the concordance rate in monozygotic twins is less than unity. One class of agents that have garnered attention in the ASD field is heavy metals. Heavy metals are known to be neurotoxins, prompting research in their possible role in the development of ASD [117-121]. Adams et al. measured the levels of 39 metals and minerals in the hair of children with ASD and a subset of their mothers as well as healthy children and a subset of their mothers [121].Using inductively coupled plasma-mass spectrometry (ICP-MS), no statistical differences in heavy metal concentrations were found in children or their mothers; however, statistical differences in the following essential minerals were found in ASD children: 45% lower iodine levels (p=0.005), 12% lower phosphorus (p=0.001).Mothers of children with ASD showed lithium levels 40% lower than mothers of healthy children (p=0.05) while mothers of ASD children aged 3-8 years had lithium levels 56% lower (p=0.005).Mercury, specifically, is widely suspected to play at least some role in the development of ASD. When looking specifically at mercury levels in the first baby haircuts of children with ASD, Holmes et al. noted a trend of lower mercury levels in the hair of autistic children (n=94) vs. controls (n=45) and potential correlations to mercury exposure in mothers. ASD child hair saw levels at 0.47 ppm compared to 3.63 ppm in controls (p<0.0000004); mercury levels varied depending on the severity of ASD and males more commonly saw lower levels (Table 7).
Table 7: Variances among individuals with ASD with respect to hair mercury levels.
Hair Mercury Level (ppm) | Percent Developmental Regression | Male: Female Ratio | |
Mild (n=27) | 0.71a | 100 | 12:15 |
Moderate (n=43) | 0.46b | 93 | 37:6 |
Severe (n=24) | 0.21c | 21 | 23:1 |
bStatistically different from mildly autistic group (p<0.0004).
cStatistically different from moderately autistic group (p<0.00002).
Mothers pregnant with a child who later developed ASD were exposed to higher levels of mercury during pregnancy than were normal controls, receiving on average 0.53 ±0.67 mercury-containing Rho D immunoglobulin shots vs. 0.09 ±29 in controls (p<0.0000004) and had 8.35 ±3.43 mercury amalgam fillings during pregnancy vs. 6.60 ±3.55 in controls p<0.1). These results suggest that autistic infants have a reduced capacity to excrete mercury through hair, despite often being subjected to higher levels of mercury than controls before birth, and so lack much of the ability of normal children to defend against the toxicity of mercury [122].
An interesting interaction between mercury ions and bilirubin oxidase, noted by Sekia et al., is a dramatic increase in enzyme activity, while the addition of cupric, cobalt, and nickel ions resulted in strong to complete enzyme inhibition. Spectroscopic measurement exhibits bilirubin oxidase activity only increased with the addition of mercury in the presence of bilirubin and bilirubin oxidase by forming a complex with bilirubin that is a more suitable substrate for bilirubin oxidase [123]. This is of special interest to our group because bilirubin is a precursor for stercobilin, which we are investigating as a potential ASD biomarker as discussed above.
Abnormal copper and zinc levels have both been shown to have effects on neurological health [124-128]. Low levels of zinc, an antioxidant, has been shown to be associated with DNA damage and repair as well as oxidative stress [125]. Copper, a redox-active element, has also been linked to conditions such as Wilson’s disease [126] and autism [127]. Faber et al. examined plasma levels of zinc, copper, and the zinc/copper ratio and found that the zinc to copper ratio in ASD subjects is below that of the bottom 2.5% of healthy children; theyspeculated that mercury toxicity might play a role in that depletion [129]. Russo et al. probed the relationship of plasma copper and zinc concentrations with ASD symptom severity, finding that autistic individuals showed lower zinc levels and significantly higher copper levels compared to controls (p=0.03); selected ASD symptom severity correlates with copper/zinc. It was hypothesized that irregular copperand zinc levels may change transmitter concentration by modulation of GABA receptors [130].
Metals may certainly provide insights into the cause and effect relationship of oxidative stress and protein regulation as related to ASD, though metal levels may be difficult to use as biomarkers because abnormal levels can be due to a number of disease states as well as environmental and nutritional factors. Despite this, when combined with other genomic, proteomic, and metabolomic information, metal measurements remain a useful tool in biomarker research.
Concluding Remarks
While the mystery of ASD etiology still remains, advances in genomics during the past decade have illuminated instances of single gene or chromosome abnormalities which lead to ASD. In addition, genomics arrays have revealed that CNVs are not uncommon in ASD. The interplay of multiple genes in the susceptibility to ASD is suspected, but specifically which particular combinations of genes are still largely unknown. ASD are therefore heterogeneous at the genomic level as they are at the phenotypic level. Thus, it appears unlikely that a small subset of genes will prove to be diagnostic for ASD. Nevertheless, genomics is already yielding clues that point to other molecular markers in the proteome and metabolome which may be far more general for ASD. In particular, markers of oxidative stress show promise as potential biomarkers of ASD. Ultimately, in spite of the heterogeneity of the disorder, protein and/or metabolite markers may eventually be found that can be useful in clinical diagnosis of ASD. Finally, the roles of environmental factors of ASD is still in its relative infancy, yet fruitful analysis in the past decade have indicated abnormalities in metal levels in ASD subjects vs. controls. How the metal levels correlate with genomic, proteomic, and metabolomics markers will certainly be an area for exploration over the next several years.Acknowledgments
We are indebted to our co-workers over the years who have worked with us on the ASD biomarker problem: Giuseppe Fanciulli, Christopher Pennington, Yong-Seok Choi, Craig Dufresne, Charmion Cruickshank, and Kevin Quinn. Early funding for this work was obtained from the United Kingdom Legal Services Commission.References
- Kanner L (1943) Autistic disturbances of affective contact. Nerv Child 2: 217-250.
- Kanner L, Eisenberg L (1957) Early infantile autism, 1943-1955. Psychiatr Res Rep Am Psychiatr Assoc: 55-65.
- Lord C, Cook EH, Leventhal BL, Amaral DG (2000) Autism spectrum disorders. Neuron 28: 355-363.
- Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, et al. (2000) The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30: 205-223.
- Eigsti IM, Shapiro T (2003) A systems neuroscience approach to autism: biological, cognitive, and clinical perspectives. Ment Retard Dev Disabil Res Rev 9: 205-215.
- Bolte S, Poustka F (2002) The relation between general cognitive level and adaptive behavior domains in individuals with autism with and without comorbid mental retardation. Child Psychiatry Hum Dev 33: 165-172.
- Herbert MR, Russo JP, Yang S, Roohi J, Blaxill M, et al. (2006) Autism and environmental genomics. Neurotoxicology 27: 671-684.
- Eisenberg L, Kanner L (1956) Childhood schizophrenia; symposium, 1955. VI. Early infantile autism, 1943-55. Am J Orthopsychiatry 26: 556-566.
- Lainhart JE, Ozonoff S, Coon H, Krasny L, Dinh E, et al. (2002) Autism, regression, and the broader autism phenotype. Am J Med Genet 113: 231- 237.
- Lord C, Shulman C, DiLavore P (2004) Regression and word loss in autistic spectrum disorders. J Child Psychol Psychiatry 45: 936-955.
- Luyster R, Richler J, Risi S, Hsu WL, Dawson G, et al. (2005) Early regression in social communication in autism spectrum disorders: a CPEA Study. Dev Neuropsychol 27: 311-336.
- Maestro S, Casella C, Milone A, Muratori F, Palacio-Espasa F (1999) Study of the onset of autism through home movies. Psychopathology 32: 292-300.
- Maestro S, Muratori F, Cesari A, Pecini C, Apicella F, et al. (2006) A view to regressive autism through home movies. Is early development really normal?. Acta Psychiatr Scand 113: 68-72.
- Filipek PA, Accardo PJ, Ashwal S, Baranek GT, Cook EH, Jr, et al. (2000) Practice parameter: screening and diagnosis of autism: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society. Neurology 55: 468-479.
- Volkmar FR, Klin A, Siegel B, Szatmari P, Lord C, et al. (1994) Field trial for autistic disorder in DSM-IV. Am J Psychiatry 151: 1361-1367.
- Willemsen-Swinkels SH, Buitelaar JK (2002) The autistic spectrum: subgroups, boundaries, and treatment. Psychiatr Clin North Am 25: 811-836.
- Bhaumik S, Branford D, McGrother C, Thorp C (1997) Autistic traits in adults with learning disabilities. Br J Psychiatry 170: 502-506.
- Vig S, Jedrysek E (1999) Autistic features in young children with significant cognitive impairment: autism or mental retardation?. J Autism Dev Disord 29: 235-248.
- DeStefano F, Chen RT (2001) Autism and measles-mumps-rubella vaccination: controversy laid to rest? CNS Drugs 15: 831-837.
- Fombonne E (1996) Is the prevalence of autism increasing?. J Autism Dev Disord 26: 673-676.
- Wing L, Potter D (2002) The epidemiology of autistic spectrum disorders: is the prevalence rising?. Ment Retard Dev Disabil Res Rev 8: 151-161.
- Yazbak FE, Lang-Radosh KL (2001) Increasing incidence of autism. Adverse Drug React Toxicol Rev 20: 60-63.
- Blumberg SJ, Bramlett MD, Kogan MD, Schieve LA, Jones JR, et al. (2013) Changes in Prevalence of Parent-reported Autism Spectrum Disorder in School-aged U.S. Children: 2007 to 2011–2012. Natl Health Stat Rep 65: 1-12.
- Butter EM, Wynn J, Mulick JA (2003) Early intervention critical to autism treatment. Pediatr Ann 32: 677-684.
- Francis K (2005) Autism interventions: a critical update. Dev Med Child Neurol 47: 493-499.
- McConnell SR (2002) Interventions to facilitate social interaction for young children with autism: review of available research and recommendations for educational intervention and future research. J Autism Dev Disord 32: 351-372.
- Ozonoff S, Goodlin-Jones BL, Solomon M (2005) Evidence-based assessment of autism spectrum disorders in children and adolescents. J Clin Child Adolesc Psychol 34: 523-540.
- Ganz ML (2007) The lifetime distribution of the incremental societal costs of autism. Arch Pediatr Adolesc Med 161: 343-349.
- Persico AM, Bourgeron T (2006) Searching for ways out of the autism maze: genetic, epigenetic and environmental clues. Trends Neurosci 29: 349-358.
- Trottier G, Srivastava L, Walker CD (1999) Etiology of infantile autism: a review of recent advances in genetic and neurobiological research. J Psychiatry Neurosci 24: 103-115.
- Dettmer K, Aronov PA, Hammock BD (2007) Mass spectrometry-based metabolomics. Mass Spectrom Rev 26: 51-78.
- Folstein S, Rutter M (1977) Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry 18: 297-321.
- Ritvo ER, Spence MA, Freeman BJ, Mason-Brothers A, Mo A, et al. (1985) Evidence for autosomal recessive inheritance in 46 families with multiple incidences of autism. Am J Psychiatry 142: 187-192.
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, et al. (1989) A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry 30: 405-416.
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, et al. (1995) Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med 25: 63-77.
- Rosenberg RE, Law JK, Yenokyan G, McGready J, Kaufmann WE, et al. (2009) Characteristics and concordance of autism spectrum disorders among 277 twin pairs. Arch Pediatr Adolesc Med 163: 907-914.
- Le Couteur A, Bailey A, Goode S, Pickles A, Robertson S, et al. (1996) A broader phenotype of autism: the clinical spectrum in twins. J Child Psychol Psychiatry 37: 785-801.
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, et al. (2011) Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry 68: 1095-1102.
- Spence SJ (2004) The genetics of autism. Semin Pediatr Neurol 11: 196-204.
- Bolton P, Macdonald H, Pickles A, Rios P, Goode S, et al. (1994) A casecontrol family history study of autism. J Child Psychol Psychiatry 35: 877-900.
- Constantino JN, Zhang Y, Frazier T, Abbacchi AM, Law P (2010) Sibling recurrence and the genetic epidemiology of autism. Am J Psychiatry 167: 1349-1356.
- Constantino JN, Todorov A, Hilton C, Law P, Zhang Y, et al. (2013) Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol Psychiatry 18: 137-138.
- Bailey A, Phillips W, Rutter M (1996) Autism: towards an integration of clinical, genetic, neuropsychological, and neurobiological perspectives. J Child Psychol Psychiatry 37: 89-126.
- Folstein SE, Rosen-Sheidley B (2001) Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet 2: 943-955.
- Skaar DA, Shao Y, Haines JL, Stenger JE, Jaworski J, et al. (2005) Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry 10: 563- 571.
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, et al. (2008) Identifying autism loci and genes by tracing recent shared ancestry. Science 321: 218- 223.
- Geschwind DH (2011) Genetics of autism spectrum disorders. Trends Cogn Sci 15: 409-416.
- Cohen D, Pichard N, Tordjman S, Baumann C, Burglen L, et al. (2005) Specific genetic disorders and autism: clinical contribution towards their identification. J Autism Dev Disord 35: 103-116.
- Schaaf CP, Zoghbi HY (2011) Solving the autism puzzle a few pieces at a time. Neuron 70: 806-808.
- Miles JH (2011) Autism spectrum disorders--a genetics review. Genet Med 13: 278-294.
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246-250.
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285-299.
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, et al. (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488: 471-475.
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82: 477-488.
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. (2007) Strong association of de novo copy number mutations with autism. Science 316: 445-449.
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, et al. (2010) Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466: 368-372.
- Beaudet AL (2007) Autism: highly heritable but not inherited. Nat Med 13: 534-536.
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, et al. (2010) Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466: 368-372.
- Weiss LA, Shen YP, Korn JM, Arking DE, Miller DT, et al. (2008) Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med358: 667-675.
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, et al. (2011) Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70: 898-907.
- Chahrour MH, Yu TW, Lim ET, Ataman B, Coulter ME, et al. (2012) Whole- Exome Sequencing and Homozygosity Analysis Implicate Depolarization- Regulated Neuronal Genes in Autism. PLoS Genet 8: 236-244.
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459: 569-573.
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70: 863-885.
- Fombonne E (2003) Epidemiological surveys of autism and other pervasive developmental disorders: an update. J Autism Dev Disord 33: 365-382.
- Robinson EB, Lichtenstein P, Anckarsater H, Happe F, Ronald A (2013) Examining and interpreting the female protective effect against autistic behavior. Proc Natl Acad Sci U S A 110: 5258-5262.
- Manzi B, Loizzo AL, Giana G, Curatolo P (2008) Autism and metabolic diseases. J Child Neurol 23: 307-314.
- Rostami A, Kalantari S, Moravej Farshi H, Zali A, Zali H, et al. (2009) Biomarkers detection of basal cell carcinoma. Pizhuhandah 14: 137Persian- 141Persian.
- Park EC, Kim G-H, Yun S-H, Lim HL, Hong Y, et al. (2012) Analysis of the endoplasmic reticulum subproteome in the livers of type 2 diabetic mice. Int J Mol Sci 13: 17230-17243.
- Rapoport SI, Nelson PT (2011) Biomarkers and evolution in Alzheimer disease. Prog Neurobiol 95: 510-513.
- Ray B, Long JM, Sokol DK, Lahiri DK (2011) Increased secreted amyloid precursor protein-α (sAPPα) in severe autism: proposal of a specific, anabolic pathway and putative biomarker. PLoS One 6: e20405.
- Lakshmi Priya MD, Geetha A (2011) A biochemical study on the level of proteins and their percentage of nitration in the hair and nail of autistic children. Clin Chim Acta 412: 1036-1042.
- Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A (2008) HMGB1: endogenous danger signaling. Mol Med 14: 476-484.
- Kim JB, Lim CM, Yu YM, Lee JK (2008) Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J Neurosci Res 86: 1125-1131.
- Emanuele E, Boso M, Brondino N, Pietra S, Barale F, et al. (2010) Increased serum levels of high mobility group box 1 protein in patients with autistic disorder. Prog Neuro-Psychopharmacol Biol Psychiatry 34: 681-683.
- Brielmaier J, Matteson PG, Silverman JL, Senerth JM, Kelly S, et al. (2012) Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One 7: e40914.
- Brune CW, Korvatska E, Allen-Brady K, Cook EH, Jr., Dawson G, et al. (2008) Heterogeneous association between engrailed-2 and autism in the CPEA network. Am J Med Genet, Part B 147B: 187-193.
- Benayed R, Choi J, Matteson Paul G, Gharani N, Kamdar S, et al. (2009) Autism-associated haplotype affects the regulation of the homeobox gene, ENGRAILED 2. Biol Psychiatry 66: 911-917.
- Yang P, Shu B-C, Hallmayer Joachim F, Lung F-W (2010) Intronic single nucleotide polymorphisms of engrailed homeobox 2 modulate the disease vulnerability of autism in a han chinese population. Neuropsychobiology 62: 104-115.
- Corbett BA, Kantor AB, Schulman H, Walker WL, Lit L, et al. (2007) A proteomic study of serum from children with autism showing differential expression of apolipoproteins and complement proteins. Mol Psychiatry 12: 292-306.
- Spector A (2000) Review: oxidative stress and disease. J Ocul Pharmacol Ther 16: 193-201.
- Sies H (1999) Glutathione and its role in cellular functions. Free Radic Biol Med 27: 916-921.
- Main PA, Angley MT, O’Doherty CE, Thomas P, Fenech M (2012) The potential role of the antioxidant and detoxification properties of glutathione in autism spectrum disorders: a systematic review and meta-analysis. Nutr Metab (Lond) 9: 35.
- Meister A, Anderson ME (1983) Glutathione. Annu Rev Biochem 52: 711- 760.
- Abdalla DS, Monteiro HP, Oliveira JA, Bechara EJ (1986) Activities of superoxide dismutase and glutathione peroxidase in schizophrenic and manic-depressive patients. Clin Chem 32: 805-807.
- Damodaran LPM, Arumugam G (2011) Urinary oxidative stress markers in children with autism. Redox Rep 16: 216-222.
- Ono H, Sakamoto A, Sakura N (2001) Plasma total glutathione concentrations in healthy pediatric and adult subjects. Clin Chim Acta 312: 227-229.
- Al-Yafee YA, Al-Ayadhi LY, Haq SH, El-Ansary AK (2011) Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol 11: 139.
- Meguid NA, Dardir AA, Abdel-Raouf ER, Hashish A (2011) Evaluation of Oxidative Stress in Autism: Defective Antioxidant Enzymes and Increased Lipid Peroxidation. Biol Trace Elem Res 143: 58-65.
- Yorbik O, Sayal A, Akay C, Akbiyik DI, Sohmen T (2002) Investigation of antioxidant enzymes in children with autistic disorder. Prostaglandins Leukot. Essent. Fatty Acids 67: 341-343.
- Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, et al. (2011) Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr Metab (Lond) 8: 34.
- James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, et al. (2004) Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr 80: 1611-1617.
- James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, et al. (2006) Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet. 141B: 947-956.
- Melnyk S, Fuchs GJ, Schulz E, Lopez M, Kahler SG, et al. (2012) Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J Autism Dev Disord 42: 367-377.
- Chauhan A, Audhya T, Chauhan V (2012) Brain Region-Specific Glutathione Redox Imbalance in Autism. Neurochem Res 37: 1681-1689.
- Sogut S, Zoroglu SS, Ozyurt H, Yilmaz HR, Ozugurlu F, et al. (2003) Changes in nitric oxide levels and antioxidant enzyme activities may have a role in the pathophysiological mechanisms involved in autism. Clin Chim Acta 331: 111- 117.
- Nelson DL, Cox MM (2000) Lehninger Principles of Biochemistry. New York: Worth Publisher.
- Sweeten TL, Posey DJ, Shankar S, McDougle CJ (2004) High nitric oxide production in autistic disorder: a possible role for interferon-γ. Biol Psychiatry 55: 434-437.
- Cohen Brett I (2004) ‘Gamma-aminobutyric Acid (GABA) and Methylmalonic Acid- The Connection with Infantile Autism’. Trends in Autism Research. Hauppauge: Nova Science Publishers, Inc. pp. 177-186.
- Cohen BI (2006) Ammonia (NH3), nitric oxide (NO) and nitrous oxide (N2O)-the connection with infantile autism. Autism 10: 221-223.
- Cohen Brett I (2002) Use of a GABA-transaminase agonist for treatment of infantile autism. Med Hypotheses 59: 115-116.
- Mayer B, Pfieffer S, Schrammel A, Koesling D, Schmidt K, et al. (1998) A new pathway of nitric oxide/cyclic GMP signaling involving S-nitrosoglutathione. J Biol Chem 273: 3264-3270.
- Seneff S, Lauritzen A, Davidson RM, Lentz-Marino L (2013) Is encephalopathy a mechanism to renew sulfate in autism?. Entropy 15: 372-406.
- Zoroglu SS, Yurekli M, Meram I, Sogut S, Tutkun H, et al. (2003) Pathophysiological role of nitric oxide and adrenomedullin in autism. Cell Biochem Funct 21: 55-60.
- Gutteridge JMC (1995) Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 41: 1819-1828.
- Chauhan A, Chauhan V, Brown WT, Cohen I (2004) Oxidative stress in autism: increased lipid peroxidation and reduced serum levels of ceruloplasmin and transferrin - the antioxidant proteins. Life Sci 75: 2539- 2549.
- Panksepp J (1979) Neurochemical Theory of Autism. Trends in Neurosciences 2: 174-177.
- Reichelt KL, Knivsberg AM (2003) Can the pathophysiology of autism be explained by the nature of discovered urine peptides?. Nutr Neurosci 6: 19- 28.
- Knivsberg AM, Reichelt KL, Hoien T, Nodland M (2002) A randomised, controlled study of dietary intervention in autistic syndromes. Nutr Neurosci 5: 251-261.
- Fukudome S, Jinsmaa Y, Matsukawa T, Sasaki R, Yoshikawa M (1997) Release of opioid peptides, gluten exorphins by the action of pancreatic elastase. FEBS Lett 412: 475-479.
- Fanciulli G, Azara E, Wood TD, Delitala G, Marchetti M (2007) Liquid chromatography-mass spectrometry assay for quantification of Gluten Exorphin B5 in cerebrospinal fluid. J Chromatogr B Analyt Technol Biomed Life Sci 852: 485-490.
- Fanciulli G, Azara E, Wood TD, Dettorl A, Delitala G, et al. (2006) Quantification of Gluten Exorphin A5 in cerebrospinal fluid by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 833: 204-209.
- Pennington CL, Dufresne CP, Fanciulli G, Wood TD (2007) Detection of gluten exorphin B4 and B5 in human blood by liquid chromatography-mass spectrometry/mass spectrometry. Open Spectrosc J 1: 9-16.
- Wood TD, Pennington CL, Choi YS (2007) Stercobilin: A Possible Biomarker for Autism? 55th American Society for Mass Spectrometry Annual Conference. Indianapolis, IN.
- Kay IT, Weiner M, Watson CJ (1963) The formation in vitro of (±)-stercobilin from bilirubin. J Biol Chem 238: 1122-1123.
- Kay IT, Weimer M, Watson CJ (1963) The formation in vitro of stercobilin from bilirubin. J Biol Chem 238: 1122-1123.
- Tell G, Gustincich S (2009) Redox state, oxidative stress, and molecular mechanisms of protective and toxic effects of bilirubin on cells. Curr Pharm Des 15: 2908-2914.
- Al-Farsi YM, Waly MI, Al-Sharbati MM, Al-Shafaee MA, Al-Farsi OA, et al. (2013) Levels of heavy metals and essential minerals in hair samples of children with autism in Oman: a case-control study. Biol Trace Elem Res 151: 181-186.
- Kern JK, Geier DA, Adams JB, Geier MR (2010) A biomarker of mercury body-burden correlated with diagnostic domain specific clinical symptoms of autism spectrum disorder. Biometals 23: 1043-1051.
- Youn SI, Jin SH, Kim SH, Lim S (2010) Porphyrinuria in Korean children with autism: correlation with oxidative stress. J Toxicol Environ Health A 73: 701-710.
- Geier DA, Kern JK, King PG, Sykes LK, Geier MR (2012) Hair toxic metal concentrations and autism spectrum disorder severity in young children. Int J Environ Res Public Health 9: 4486-4497.
- Adams JB, Holloway CE, George F, Quig D (2006) Analyses of toxic metals and essential minerals in the hair of Arizona children with autism and associated conditions, and their mothers. Biol Trace Elem Res 110: 193- 209.
- Holmes AS, Blaxill MF, Haley BE (2003) Reduced levels of mercury in first baby haircuts of autistic children. Int J Toxicol 22: 277-285.
- Seki Y, Takeguchi M, Murao S, Shin T, Okura I (1995) Abnormal effect of mercury ions on bilirubin oxidase activity. J Mol Catal A Chem 96: L11-L13.
- Song Y, Leonard SW, Traber MG, Ho E (2009) Zinc deficiency affects DNA damage, oxidative stress, antioxidant defenses, and DNA repair in rats. J Nutr 139: 1626-1631.
- Powell SR (2000) The antioxidant properties of zinc. J Nutr 130: 1447S-1454S.
- Das SK, Ray K (2006) Wilson’s disease: an update. Nat Clin Pract Neurol 2: 482-493.
- Chauhan A, Sheikh AM, Chauhan V (2008) Increased copper-mediated oxidation of membrane phosphatidylethanolamine in autism. Am J Biochem Biotechnol 4: 95-100.
- Halatek T, Lutz P, Stetkiewicz J, Krajnow A, Wieczorek E, et al. (2013) Comparison of neurobehavioral and biochemical effects in rats exposed to dusts from copper smelter plant at different locations. J Environ Sci Health A Tox Hazard Subst Environ Eng 48: 1000-1011.
- Faber S, Zinn GM, Kern JC 2nd, Kingston HM (2009) The plasma zinc/serum copper ratio as a biomarker in children with autism spectrum disorders. Biomarkers 14: 171-180.
- Russo AJ, Bazin AP, Bigega R, Carlson RS III, Cole MG, et al. (2012) Plasma copper and zinc concentration in individuals with autism correlate with selected symptom severity. Nutr Metab Insights 5: 41-47.