
Journal of Neurology and Psychology
Download PDF
Research Article
*Address for Correspondence: Hassan Al-Jafar, Department of Haematology, Amiri Hospital, Gulf street, Kuwait city, Kuwait, E-mail: cbc9@Hotmail.com
Citation: Al-Jafar H, Hashem K, Alhasan AA, Lamdhade S, AlDallal S, et al. Moyamoya Disease: A Rare Sickle Cell Trait Neurological Complication. J Neurol Psychol. 2016;4(1): 3.
Copyright © 2016 Al-Jafar H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 4, Issue: 1
Submission: 17 November 2015 | Accepted: 21 January 2016 | Published: 26 January 2016
Reviewed & Approved by: Dr. Jennifer Larimore, Department of Biology and Neuroscience Program, Agnes Scott College, USA


Moyamoya Disease: A Rare Sickle Cell Trait Neurological Complication
Hassan Al-Jafar1*, Khaled Hashem2, Ali Abo Alhasan3, Shashishekhar Lamdhade4, Salma AlDallal1, and Fayza Abdulla Alhajri2
- 1Department of Haematology, Amiri Hospital, Gulf street, Kuwait City, Kuwait
- 2Department of Radiology, Amiri Hospital, Gulf street, Kuwait City, Kuwait
- 3Department of Neurosurgery, Ibn Sina Hospital, Sabah Medical Region, Kuwait
- 4Department of Neurology, Amiri Hospital, Gulf street, Kuwait City, Kuwait
*Address for Correspondence: Hassan Al-Jafar, Department of Haematology, Amiri Hospital, Gulf street, Kuwait city, Kuwait, E-mail: cbc9@Hotmail.com
Citation: Al-Jafar H, Hashem K, Alhasan AA, Lamdhade S, AlDallal S, et al. Moyamoya Disease: A Rare Sickle Cell Trait Neurological Complication. J Neurol Psychol. 2016;4(1): 3.
Copyright © 2016 Al-Jafar H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology and Psychology | ISSN: 2332-3469 | Volume: 4, Issue: 1
Submission: 17 November 2015 | Accepted: 21 January 2016 | Published: 26 January 2016
Reviewed & Approved by: Dr. Jennifer Larimore, Department of Biology and Neuroscience Program, Agnes Scott College, USA
Abstract
Introduction: Sickle cell disease is an inherited blood disorder with multisystem complications, including cerebrovascular accidents primarily due to micro infarcts. However, people with sickle cell trait, the heterozygous form of sickle cell disease, usually lead a normal, healthy life. Very rarely, severe tissue hypoxia, acidosis, dehydration, and hypothermia in sickle cell trait increase red blood cell polymerization and sickling, which could lead to micro infarcts in blood vessels. Moyamoya disease is a rare neurovascular complication caused by repeated strokes after arterial cerebral infarction. It commonly manifests as ischemic stroke in young patients, but mainly as intracranial hemorrhage in adults.Case presentation: A 14-year-old girl with sickle cell trait, who had been living a normal and healthy life, started to complain of severe persisting headache, migraine, drowsiness, and a tendency to sleep. Brain magnetic resonance imaging and angiography revealed moyamoya disease. Hematological treatment with hydroxyurea, aspirin, and an NSAID was commenced for this patient.
Conclusion: Under rare circumstances, sickle cell trait could lead to sickle cell disease complications. Although moyamoya has been seen in sickle cell disease, it is a very rare neurological complication of sickle cell trait. To the best of our knowledge, only three cases of moyamoya have been reported in individuals with sickle cell trait.
Keywords
Moyamoya; Rare; Sickle cell trait; Neurological complicationIntroduction
Sickle cell disease (SCD) is a hereditary autosomal recessive blood disorder. It is the most frequent type of hemoglobinopathy. Patients with SCD present with a broad range of phenotypic hemoglobin structures; they vary clinically from mild vaso-occlusive crises to severe clinical manifestations [1]. Neurological dysfunction is a wellknown complication in patients with SCD. It usually manifests as an acute cerebrovascular accident or as a headache, seizure, or cognitive decline [2]. Children and teenagers are particularly vulnerable toovert cerebrovascular complications of SCD [3]. Sickle cell trait (SCT) is the heterozygous form of SCD, found in approximately 300 million people. Traditionally, SCT has been viewed as a benign condition, a non-disease status, without any painful episodes that are characteristic of homozygous SCD. On a population basis, SCT has no discernible impact on life expectancy. Hemoglobin and hematocrit values in such individuals are similar to those of persons without hemoglobinopathy. People with SCT are eligible for blood donation in the United States and other countries. SCT is rarely accompanied by severe tissue hypoxia, acidosis, dehydration, and hypothermia, which may increase red blood cell (RBC) polymerization; this can lead to RBC sickling, which can cause micro infarcts in capillary blood vessels [4]. Moyamoya disease (MM) is a chronic cerebral vasculopathy first described in 1957 by Takeuchi and Shimizu from Japan [5]. Moyamoya means hazy in Japanese. This disease derived its unusual name from the angiographic appearance of cerebral vesselsresembling a ‘puff of smoke’ [6]. MM is characterized by slowly progressive stenosis and obliteration of the large vessels at the base of the brain, mainly affecting the supraglenoid segment of the internal carotid artery with the initial portion of the anterior or middle cerebral arteries and the posterior cerebral arteries [7]. Also, it is associated with collateral vessels at the base of the brain (moyamoya vessels) [8]. However, the diagnostic criteria for this disease are mainly based on angiographic findings [9]. Children and adults with MM may have different clinical presentations; the symptoms and clinical course vary widely, ranging from being asymptomatic to manifesting as severe neurologic deficits. Children more commonly have ischemic events and may experience hemiparesis, monoparesis, sensory impairment, involuntary movements, headaches, dizziness, seizures, mental retardation, or persistent neurologic deficits. In adults, symptoms and signs are similar to those in children, but sudden onset intraventricular, subarachnoid, or intracerebral hemorrhage is more common [10]. MM, in general, is a rare neurovascular disorder [11]. To our knowledge, only three cases of MM have been reported in patients with SCT [12].Case Presentation
A 14-year-old girl, who had been living a completely normal life, started to complain of a headache, migraine, drowsiness, and a tendency to sleep. There was no family history of MM. A complete blood count revealed microcytic hypochromic RBCs. Highperformance liquid chromatography showed double heterozygosity of SCT and β-thalassemia trait. Laboratory test results were as follows: white blood cell count, 7.8×109 /L (normal 4-10×109 /L); RBC count, 5.19 (normal 4-5.2×1012/ L); hemoglobin level, 12.5 (normal 12.0-15.0) g/L and platelet count,445×109 /L (normal 170-450×109 /L). High-performance liquid chromatography results were as follows: HbA 63.02% (normal 96-98%); HbF < 1% (normal < 1%); HbA2 3.79% (normal 1.5-3.5%) and HbS 32.89%; liver function tests revealed total protein level of 70 (normal 61-79) g/L, an albumin level of 47 (normal 35-48) g/L, an alkaline phosphatase level of 89 (normal 42-98) IU/L, an alanine transaminase level of 9 (normal 10-60) IU/L, and an aspartate aminotransferase level of 17 (normal 10-42) IU/L. Renal function tests revealed a blood urea nitrogen level of 3.8 (2.5-7.1) mmol/L and a creatinine level of 46 (normal 53-88) mmol/L. Brain magnetic resonance imaging and angiography were performed and revealed images of MM (Figures 1 and 2). The patient was prescribed topiramate tablets (Topamax) 25 mg once daily as prophylaxis for migraine and etoricoxibtablets (Arcoxia) 90 mg once daily for headache. She was referred to the hematology department where aspirin tablets 81 mg once daily and hydroxyurea capsules 500 mg once daily were added to prevent cerebral thrombosis and to increase the HbF level. Cranial neurosurgery was planned. No blood transfusion treatment was used in this case because she had SCT, and the baseline HbS was only 32.89%. Instead, treatment with hydroxyurea was commenced to increase the HbF ratio.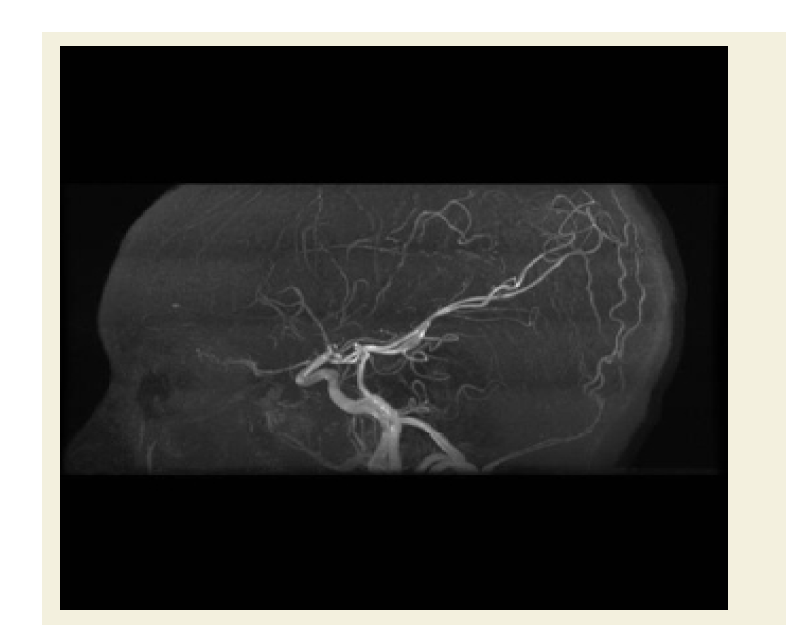
Figure 1: Sagittal MRA ToF image of the brain shows bilateral attenuated of the anterior cerebral artery and narrowed supraclinoid part of internal carotidartery.
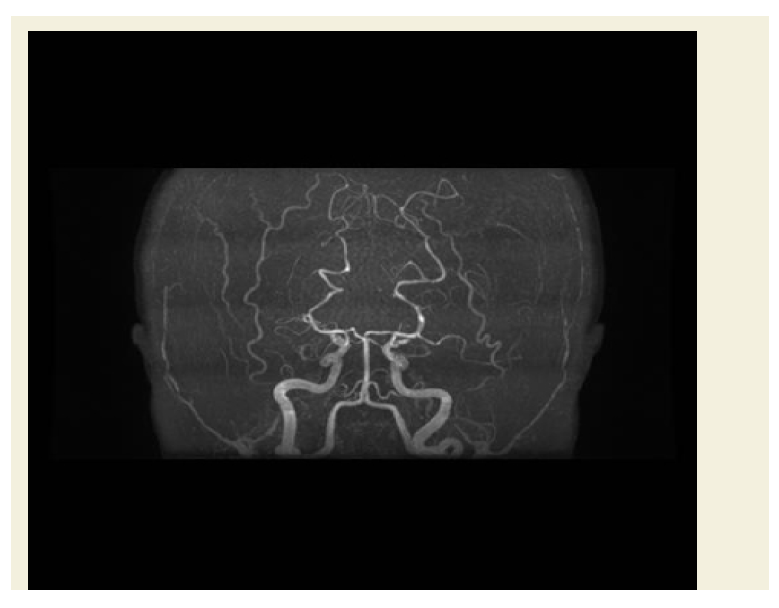
Figure 2: Coronal MRA ToF image of the brain shows bilateral attenuation of the middle cerebral arteries and supraclinoid part of both internal carotid arteries in a typical picture of Moyamoya.
Discussion
Compared with normal populations, individuals with SCT show increased coagulation activity measures with higher levels of D-dimers, thrombin-antithrombin complexes, and prothrombin fragments, and their absolute blood monocyte levels are higher [13]. Subclinical sickling occurs in these cases upon loss of normal phospholipid asymmetry, resulting in abnormal phosphatidylserine that contributes to hemostatic problems [14,15]. Moyamoya angiopathyis a well known complication of SCD but has rarely been observed in SCT or other hemoglobinopathies [16]. Patients who develop MM have a high chance of recurrent strokes like collaterals on imaging studies; these should be treated as soon as the diagnosis is confirmed [17]. Suspected patients should refer promptly to imaging studies. A large survey from Japan showed no significant differences in outcome between medically or surgically treated patients. Surgery should be considered only in high-risk patients. Antiplatelet agents have been used to prevent emboli and microthrombi formation at sites of arterial stenosis because these could be a possible cause of ischemic symptoms. Anticoagulants, such as coumadin in and lowmolecular- weight heparin, are rarely used [18]. At Children’s Hospital Boston, children with MM are treated with lifelong aspirin therapy with those younger than six years of age receive 81 mg/day and older children receive variable doses depending on the presence or absence of symptoms. Genetic analyses have suggested associations between MM patients and chromosomal abnormalities [19,20].Hydroxyurea is thought to be an effective drug for the management of SCD as it leads to a variety of alterations in RBCs, including increased HbF, mean cell volume; hydration that probably increases RBC lifespan and reduced hemoglobin polymerization. Hydroxyurea also alternate the RBC membrane and decreases expression of adhesion molecules [21]. Chronic transfusion therapy aims to maintain sickle hemoglobin levels below 30% to reduce the likelihood of strokes. However, in patients with SCD, transfusions seem to be required for long-term therapy because 50% of patients are susceptible to cerebrovascular accidents after transfusions are discontinued [22]. Medical treatment is recommended for patients with MM with acute infarction and relatively only mild MM to prevent further stroke. Antithrombotic drugs, such as aspirin, heparin, and argatroban, are recommended for adult patients presenting with cerebral infarction. Supporting treatments include controlling blood pressure levels, blood sugar levels, and pain; seizure medication may also be needed during the acute phase. For the prevention of recurrent ischemic attack in the chronic phase, aspirin is recommended. Other drugs, such as clopidogrel or thienopyridine, can be used when aspirin is not tolerated or ineffective [23]. However, there are few data from long-term studies evaluating treatment outcomes or from comparative analyses of surgical treatment in adult patients with MM [24,25]. Further study of this disorder is required to improve understanding and, ultimately, develop better therapy for MM [20].
Conclusion
People with SCT usually lead a normal life; however, rarely and under specific circumstances, they may experience SCD complications. MM in SCT is a very rare finding. It could be a mild or progressive and devastating disease, so it must be managed and followed up carefully by a hematologist, neurologist, and neurosurgeon in cooperation to achieve the best specialized care.References
- Adewoyin AS (2015) Management of sickle cell disease: a review for physician education in Nigeria (Sub-Saharan Africa). Anemia 2015: 21.
- Prengler M, Pavlakis SG, Prohovnik I, Adams RJ (2002) Sickle cell disease: the neurological complications. Ann Neurol 51: 543-552.
- Deus-Silva L, Bonilha L, Damasceno BP, Costa AL, Yasuda CL, et al. (2013) Brain perfusion impairment in neurologically asymptomatic adult patients with sickle-cell disease shown by voxel-based analysis of SPECT images. Front Neurol 4: 207.
- Kark J (2000) Sickle Cell Trait. Howard Universty School of Medicine, Center for Sickle Cell Disease, 2121 Georgia Avenue Washington, DC 20059, USA.
- Takeuchi K, Shimizu K (1957) Hypogenesis of bilateral internal carotid artery. Shinkei 9: 37-43.
- Goyal JP, Rao SS, Trivedi S (2011) Moya moya disease in a child: a case report. Case Rep Neurol Med 2011: 329738.
- Aicardi J (1988) Disease of the nervous system in childhood. Mac Keith, 2nd Edn, London, pp. 554-556.
- Shoukat S, Itrat A, Taqui AM, Zaidi M, Kamal AK (2009) Moyamoya disease: A clinical spectrum, literature review and case series from a tertiary care hospital in Pakistan. BMC Neurol 9: 15
- Ruhela M, Kumar D, Meena CB, Bagarhatta R (2014) Moyamoya disease with ebstein anomaly: an unusual co-occurrence. AJMCR 2: 111-113.
- Zipfel GJ, Fox Jr DJ, Rivet DJ (2005) Moyamoya disease in adults: the role of cerebral revascularization. Skull Base 15: 27-41.
- Adachi K, Yamamoto Y, Kameyama E, Suzuki H, Horinouchi T (2005) Early postoperative complications in patients with Moyamoya disease--a comparison of inhaled anesthesia with total intravenous anesthesia (TIVA). Masui 54: 653-657.
- Hune SY, deVeber, GA, MacGregor DL, Dirks PB, Yu HW, et al. (2007) Nursing Issues in caring for children with moyamoya vasculopathy and their families. J Neurosci Nurs 39: 33- 39
- Tsaras G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y (2009) Complications associated with sickle cell trait: a brief narrative review. Am J Med 122: 507-512.
- Austin H, Key NS, Benson JM, Lally C, Dowling NF, et al. (2007) Sickle cell trait and the risk of venous thromboembolism among blacks. Blood 110: 908-912.
- Saxena P, Dhiman P, Bihari C, Rastog A (2015) Sickle cell trait causing splanchnic venous thrombosis. Case Reports Hepatol 2015: 3.
- Brockmann K, Stolpe S, Fels C, Khan N, Kulozik AE, et al. (2005) Moyamoya syndrome associated with hemolytic anemia due to Hb Alesha. J Pediatr Hematol Oncol 27: 436-440.
- Makubi A, Soka D, Makani J (2012) Moyamoya disease, a rare cause of recurrent strokes in an African sickle cell child: does hydroxyurea have a role in this context? Int J Child Health Nutr 1: 82-85.
- Scott RM, Smith ER (2009) Moyamoya disease and moyamoya syndrome. N Engl J Med 360: 1226-1237.
- Ibrahimi DM, Tamargo RJ, Ahn ES (2010) Moyamoya disease in children. Childs Nerv Syst 26: 1297-1308
- Kim JE, Jeon JS (2014) An update on the diagnosis and treatment of adult moyamoya disease taking into consideration controversial issues. Neurol Res 36: 407-416.
- Caldarelli M, Di Rocco C, Gaglini P (2001) Surgical treatment of moyamoya disease in pediatric age. J Neurosurg Sci 45: 83-91.
- Ghatpande SS, Choudhary PK, Quinn CT, Goodman SR (2010) In vivo pharmaco-proteomic analysis of hydroxyurea induced changes in the sickle red blood cell membrane proteome. J Proteomics 73: 619-626.
- Roach ES, Golomb MR, Adams R, Biller J, Daniels S, et al. (2008) Management of stroke in infants and children. Stroke 39: 2644-2691.
- Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis, Health Labour Sciences Research Grant for Research on Measures for Infractable Diseases (2009) Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis). Neurol Med Chir (Tokyo) 52: 245-266.
- Smith ER, Scott RM (2005) Surgical management of moyamoya syndrome. Skull Base 15: 15-26