
Journal of Neurology and Psychology
Download PDF
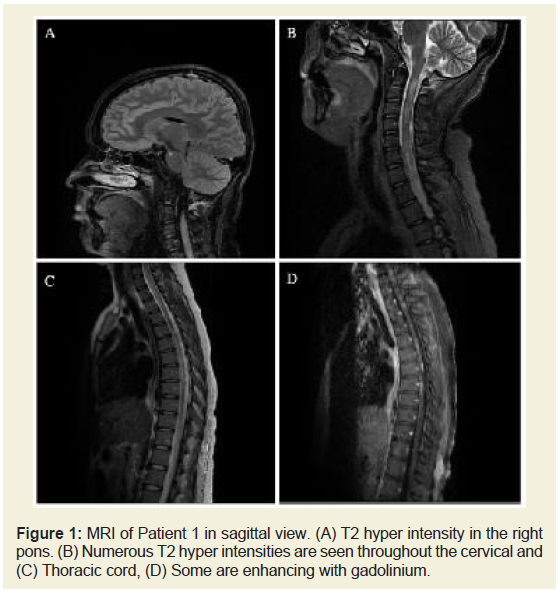 Figure 1: MRI of Patient 1 in sagittal view. (A) T2 hyper intensity in the right
pons. (B) Numerous T2 hyper intensities are seen throughout the cervical and
(C) Thoracic cord, (D) Some are enhancing with gadolinium.
Figure 1: MRI of Patient 1 in sagittal view. (A) T2 hyper intensity in the right
pons. (B) Numerous T2 hyper intensities are seen throughout the cervical and
(C) Thoracic cord, (D) Some are enhancing with gadolinium.
Case Report
Aquaporin-4 Protein Antibody Seropositivity after Acute SARS-CoV-2 Infection
Tan Y1*, Zuberi HZ2, Hernandez RM1 and Avila M1
1Department of Neurology, Texas Tech University Health Sciences
Center, Lubbock, Texas
2Texas Tech University Health Sciences Center School of Medicine,
Lubbock, Texas
*Address for Correspondence:
Tan Y, Department of Neurology, Texas Tech University Health Sciences
Center, Lubbock 79430, Texas; Telephone: 806.743.2391; E-mail:
Yuanyuan.Tan@ttuhsc.edu
Submission: August 16, 2022
Accepted: September 09, 2022
Published: September 12, 2022
Copyright: © 2022 Tan Y, et al. This is an open access article distributed
under the Creative Commons Attribution License, which permits
unrestricted use, distribution, and reproduction in any medium, provided
the original work is properly cited.
Abstract
Background:
Development of autoimmune neurological disorders
after Coronavirus Disease 2019 (COVID-19) has been reported. Though
many cases of multiple sclerosis developing after COVID-19 are
present in current literature, Neuromyelitis Optica Spectrum Disorder
(NMOSD) is much rarer sequela of the disease.
Methods:
Two cases that meet the international consensus
diagnostic criteria for NMOSD were encountered at a regional hospital
in West Texas in the same month. Both were preceded by acute
SARS-CoV-2 infection and developed newly diagnosed NMOSD with
Aquaporin-4 Protein Antibody seropositivity.
Results:
Case 1 was a 28-year-old Hispanic female who presented
with opsoclonus and ophthalmoplegia; Case 2 was a 20-year-old
African American female who presented with transverse myelitis. Both
patients had no neurological co morbidities or symptoms prior to SARSCoV-
2 infection. Neither of them was vaccinated for COVID-19, and
both were of non-Caucasian ethnicity. They presented with a typical
features including younger onset, ocular presentation of opsoclonus,
negative neuroimaging, no response to steroids, and relapse after a
short interval.
Conclusion:
New developments of NMOSD in previously healthy
individuals can be a neurological sequela of COVID-19, especially
among unvaccinated individuals. The correlation and pathophysiology
of NMOSD after COVID-19 are not fully understood, but molecular
mimicry of the virus and cytokine storm are postulated mechanisms.
Additional observational studies are needed to further explore the
correlation between acute COVID-19 infection and NMOSD.
Keywords
Neuromyelitis Optica Spectrum Disorder; Aquaporin-4 Protein
Antibody; Coronavirus Disease 2019; SARS-CoV-2
Introduction
Neuromyelitis Optica Spectrum Disorder (NMOSD) is a
rare autoimmune disorder of the central nervous system (CNS)
characterized by transverse myelitis and optic neuritis. The discovery
of Aquaporin-4 protein antibody (AQP-4 Ab) in 2004 has greatly
advanced the diagnosis of NMOSD, and its seropositivity was found
to be associated with an increased risk of recurrent attacks [1].
While neurological deficits are a common manifestation following
Corona virus Disease 2019(COVID-19) [2], The development of
autoimmune disease is less frequently depicted. There have been
several cases reporting the development of relapsing-remitting
multiple sclerosis after COVID-19 infection [3], but there is limited
literature pertaining to the prevalence of NMOSD after COVID-19,
and some of these cases are with seronegative AQP-4 Ab [4,5]. Here,
we describe two AQP-4Ab seropositive NMOSD cases that occurred
as a sequela to SARS-CoV-2 infections as well as their clinical features.
Methods/Results
Both patients met international consensus diagnostic criteria for
NMOSD and had no neurological symptoms prior to being infected
with SARS-CoV-2 [6].
Case 1:
A 28-year-old Hispanic female with a history of systemic lupus
erythematosus presented with progressive right leg paresthesia and
paresis 3 weeks after a second time COVID-19 infection without
prior vaccination. Paresthesia and weakness of the left leg, blurry
vision and dyschromatopsia of the left eye, and bladder incontinence
ensued shortly after. Physical exam revealed weakness involving
four extremities, with right worse than left, diminished right patellar
reflex, and a sensory level of T4 more pronounced on the right
side. Fundoscopy confirmed left eye papillitis. Magnetic Resonance
Imaging (MRI) was remarkable for numerous patchy enhancing
lesions throughout thecervical and thoracic spinal cord (Figure 1), a sub-centimeter lesion at the right pons, and the left optic nerve
with high FLAIR intensity. No pleocytosis and protein elevation was
present in the cerebrospinal fluid (CSF). She received a seven day
course of high dosage Solumedrol (methylprednisolone) and later
received a five day course of intravenous immunoglobin (IVIG) due
to a lack of improvement in symptoms from the steroids. The patient’s
motor strength was significantly improved after receiving IVIG.
AQP-4 Ab was found to be positive in serum. Following treatment,
the patient experienced her first relapse after one month and second
relapse after three months.
Case 2:
A 20-year-old African American female with no history of
autoimmune disease or other co morbidities presented with diplopia
as well as blurry vision and ptosis of the left eye. Her symptoms
rapidly progressed, and right eye ptosis and weakness involving all
four extremities developed within three days after admission to the
hospital. The patient reported no prodromal infection besides a mild
SARS-CoV-2 infection three months prior, which was associated
with new-onset left retro orbital headache. Her headache persisted
and became worse during her emergency room visit, and it was
described as feeling like her “left eye was going to explode.” Physical
exam showed blurry vision of the left eye, opsoclonus with oscillopsia,
bilateral ptosis, bilateral abducens nerve palsy with left globe being
worse than right, left facial paresis, and generalized weakness with
preserved reflexes. An extensive workup was performed. Full
neuro-axis MRI with and without contrast, Magnetic Resonance
Angiography (MRA) and Magnetic Resonance Venography (MRV)
of the brain, full body CT, CSF studies, myasthenia gravis autoantibodies,
autoimmune and paraneoplastic antibodies, ganglioside
auto-antibodies, common tumor markers, and muscle enzymes were
all negative. The patient was treated with high doses of Solumedrol
but was later switched to IVIG treatment due to a lack of symptom
improvement. IVIG was discontinued due to dyspnea during infusion.
Plasmapheresis was given and her symptoms stabilized. Serum AQP-
4 Ab returned positive. One month later, she experienced an attack
of complete left eye achromatopsia and describedseeing everything
as yellow. She was evaluated by a neuro-ophthalmologist who agreed
with the diagnosis of NMOSD and considered this could be a lesser
common variant: acute brainstem syndrome. Treatment with highdose
steroids again failed to provide any improvement. A repeat
full neuro-axis MRI was unremarkable. Nerve conduction studies
and needle electromyography showed no signs of neuropathy and
myopathy.
Discussion
The two patients described had similarities in case presentation:
they both had minor COVID-19 symptoms that did not require
hospitalization or supplemental oxygen and were healthy individuals
who subsequently developed disabling neurological presentations
after recovering from COVID-19. Additionally, they were both young
females of non-Caucasian ethnicity (Hispanic and African American)
and unvaccinated status. Both patients had unremarkable CSF results
and a poor response to steroids.
NMOSD is known to cause recurrent attacks; however, the
interval between attacks in our patients was much shorter compared
to general NMOSD patients (8-12 months) [7]. Atypical features were also noted, including opsoclonus and complete unremarkable MRI in
the second patient. Brainstem involvement is now more commonly
discovered due to the broadened criteria of NMOSD. Brain MRI
can be normal at initial presentation, and the lesions in brainstem
are usually less extensive compared to spinal cord. Cases with small
dot-like lesions at brainstem have been reported [8]. The atypical
features of our patients and short interval to recurrent attacks suggest
a low threshold for post-COVID-19AQP-4 Ab testing and NMOSD
evaluation, so timely management can be pursued.
The correlation of NMOSD after acute COVID-19 infection is not
fully understood. Demyelinating changes may occur due to a hyper
inflammatory state with release of cytokines caused by infection,
leading to glial activation, or it may occur as part of a delayed
immune response [9]. Molecular mimicry of SARS-CoV2 antigens
and neurological self-antigens is another potential mechanism. It was
reported that both natural SARS-CoV-2 infection and SARS-CoV-2
vaccination can induce the formation of AQP-4 Ab and lead to newly
diagnosed NMOSD, or it can also trigger a relapse in patients who
already had an established diagnosis [5,10-13]. Of note, the postimmunization
NMOSD cases are not limited to a certain type or
certain company-made vaccine [10]. This literature suggests that
the shared antiviral immune response between natural infection and
vaccinations may play a role in the NMOSD pathogenesis.
Conclusion
Our two cases indicate NMOSD after acute COVID-19 can
have atypical presentations. AQP-4 Ab test is vital for the accurate
diagnosis and timely management of the NMOSD. The correlation
of NMOSD after acute COVID-19 is not fully understood. Molecular
mimicry of the virus and cytokine storm are postulated mechanisms
for NMOSD pathogenesis. Shared antiviral immune response
between natural infection and COVID-19 vaccination may also play
a role. Additional observational studies are needed to further explore
the correlation between acute COVID-19 infection and NMOSD.
Acknowledgment
No funding was received for this case study. The authors have
no conflicts of interest to declare or financial interest to report. All
authors approve of the final version of this manuscript and agree
to be accountable for its contents. Written informed consent was
obtained from the patients for publication of this case report along
with corresponding images.
References
Citation
Tan Y, Zuberi HZ, Hernandez RM, Avila M. Aquaporin-4 Protein Antibody Seropositivity after Acute SARS-CoV-2 Infection. J Neurol Psychol. 2022;
9(1): 07