
Journal of Orthopedics & Rheumatology
Download PDF
Review Article
*Address for Correspondence: Jun Li, Division of Clinical Immunology and Rheumatology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, 35294, USA; Tel: 205-934-8909; Fax: 205-996-6788; E-mail: junliuab@uab.edu
Citation: Li J, Hsu HC, Mountz JD. The Dynamic Duo-Inflammatory M1 macrophages and Th17 cells in Rheumatic Diseases. J Orthopedics Rheumatol. 2013;1(1): 4.
Copyright © 2013 Li J, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Orthopedics & Rheumatology | ISSN: 2334-2846 | Volume: 1, Issue: 1
Submission: 30 October 2013 | Accepted: 19 November 2013 | Published: 25 November 2013
Reviewed & Approved by: Dr. Andrew P. Andonopoulos, Department of Internal Medicine and Rheumatology, Patras University School of Medicine, Greece

The Dynamic Duo–Inflammatory M1 macrophages and Th17 cells in Rheumatic Diseases
Jun Li1*,Hui-Chen Hsu1 and John D. Mountz1,2
- 1Division of Clinical Immunology and Rheumatology, Department of Medicine, University of Alabama at Birmingham, USA
- 2Department of Medicine, Birmingham VA Medical Center, USA
*Address for Correspondence: Jun Li, Division of Clinical Immunology and Rheumatology, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, 35294, USA; Tel: 205-934-8909; Fax: 205-996-6788; E-mail: junliuab@uab.edu
Citation: Li J, Hsu HC, Mountz JD. The Dynamic Duo-Inflammatory M1 macrophages and Th17 cells in Rheumatic Diseases. J Orthopedics Rheumatol. 2013;1(1): 4.
Copyright © 2013 Li J, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Orthopedics & Rheumatology | ISSN: 2334-2846 | Volume: 1, Issue: 1
Submission: 30 October 2013 | Accepted: 19 November 2013 | Published: 25 November 2013
Reviewed & Approved by: Dr. Andrew P. Andonopoulos, Department of Internal Medicine and Rheumatology, Patras University School of Medicine, Greece
Abstract
The synovial tissue of Rheumatoid Arthritis (RA) patients is enriched with macrophages and T lymphocytes which are two central players in the pathogenesis of RA. Interaction between myeloid cells and T cells are essential for the initiation and progression of the inflammatory processes in the synovium. With the rapid evolution of our understanding of how these two cell types are involved in the regulation of immune responses, RA is emerging as an ideal disease model for investigating the cell-cell interactions and consequently introducing novel biologic agents that are designed to disrupt these processes. This review will discuss the bidirectional interaction between the IL-23+inflammatory macrophages and IL-17+ GM-CSF+CD4 T cells in rheumatic diseases as well as potential antirheumatic strategies via apoptosis induction in this context.Introduction
Bidirectional positive feedback loop between macrophages and CD4 T cellsMacrophage subpopulations are developed from monocytes as inflammatory M1 macrophages that produce Tumor Necrosis Factor-alpha (TNF-α), Interleukin (IL)-6 and IL-23, or anti-inflammatory M2 macrophages that produce IL-10 and Transformation Growth Factor (TGF)-β [1,2]. In vitro experiments can reproducibly develop such divergent macrophage subpopulations from bone marrow precursors by Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF), Lipopolysaccharide (LPS) for M1 macrophages, whereas M2 macrophages, in contrast, can be differentiated with IL-4 or M-CSF. M1 macrophage differentiation is regulated by a transcription factor IRF-5, whereas M2 macrophage is reinforced by the transcription factor IRF-4 [3,4]. CD4 T cell subpopulations likewise can be inflammatory and anti-inflammatory. Inflammatory CD4 T cells include Th1 that primarily produce IFN-γ and Th17 that predominately produce IL-17, but also IL-21, and IL-22. A transitional population of CD4 cells, known as Th1/17, express both IFN-γ and IL-17 and have been found to be the most pathogenic CD4 T cells associated with several disease types including rheumatoid arthritis [5] and Multiple Sclerosis (MS) [6,7]. The inhibitory roles of regulatory T cells in RA have also been established in both human patients and mouse models [8]. There is an interdependent and synergistic activity of M1 inflammatory macrophages and Th1/17 inflammatory CD4 T cells (Figure 1). As discussed above, M1 macrophages not only express TNF-α and IL-6, but also IL-23. IL-23 acts through the IL-23 receptor (IL-23R) expressed on CD4 T cells to promote development of Th17 T cells. The primary molecular mechanism for Th17 T cell development is up regulation of the transcription factor, RAR-Related Orphan Receptor Gamma (RORγt), after IL-23R stimulation [9-11]. This skews development of CD4 T cells away from an IFN-γ producing Th1 CD4 T cell to an IL-17 producing Th17 CD4 T cell subpopulation. Importantly, the IL-23R-RORγt signaling also promotes the development of the transitional IFN-γ and IL-17 producing CD4 T cell subpopulation, Th1/17[10], which may be derived from a Th1 population as it undergoes chromatin remodeling. Importantly, GMCSF marks the highly pathogenic Th1/17 population and can act through the GM-CSF receptor (CSF2R) expressed on macrophages to promote expression of IRF-5 and reinforces the M1 macrophage differentiation. Therefore, the co-existence of IL-23 producing M1 macrophages and GM-CSF producing Th1/17 T cells forms a positive interactive inflammatory positive feedback circuit that is associated with the highest levels of inflammation in autoimmune disease including RA[12], multiple sclerosis (MS) [11], and possibly, Systemic Lupus Erythematosus (SLE) [13].
There is an interdependent and synergistic activity of M1 inflammatory macrophages and Th1/17 inflammatory CD4 T cells (Figure 1). As discussed above, M1 macrophages not only express TNF-α and IL-6, but also IL-23. IL-23 acts through the IL-23 receptor (IL-23R) expressed on CD4 T cells to promote development of Th17 T cells. The primary molecular mechanism for Th17 T cell development is up regulation of the transcription factor, RAR-Related Orphan Receptor Gamma (RORγt), after IL-23R stimulation [9-11]. This skews development of CD4 T cells away from an IFN-γ producing Th1 CD4 T cell to an IL-17 producing Th17 CD4 T cell subpopulation. Importantly, the IL-23R-RORγt signaling also promotes the development of the transitional IFN-γ and IL-17 producing CD4 T cell subpopulation, Th1/17[10], which may be derived from a Th1 population as it undergoes chromatin remodeling. Importantly, GMCSF marks the highly pathogenic Th1/17 population and can act through the GM-CSF receptor (CSF2R) expressed on macrophages to promote expression of IRF-5 and reinforces the M1 macrophage differentiation. Therefore, the co-existence of IL-23 producing M1 macrophages and GM-CSF producing Th1/17 T cells forms a positive interactive inflammatory positive feedback circuit that is associated with the highest levels of inflammation in autoimmune disease including RA[12], multiple sclerosis (MS) [11], and possibly, Systemic Lupus Erythematosus (SLE) [13].
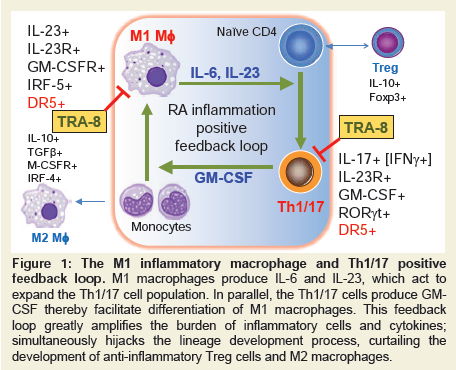
Figure 1: The M1 inflammatory macrophage and Th1/17 positive feedback loop. M1 macrophages produce IL-6 and IL-23, which act to expand the Th1/17 cell population. In parallel, the Th1/17 cells produce GM-CSF thereby facilitate differentiation of M1 macrophages. This feedback loop greatly amplifies the burden of inflammatory cells and cytokines; simultaneously hijacks the lineage development process, curtailing the development of anti-inflammatory Treg cells and M2 macrophages.
Biologic therapies of rheumatic diseases
Advanced treatment of autoimmune diseases including RA and SLE are currently approached through biologic therapies that target specific pathogenic cytokines or interactive molecules [14]. RA is primarily treated with TNF-α biologic neutralization reagents including soluble receptors to TNF-α [Etanercept (Enbrel®)] or antibodies to TNF-α [Infliximab (REMICADE®) and Adalimumab (HUMIRA®)]. Interactions between Antigen-Presenting Cells (APC) and CD4 T cells in RA have been targeted using Abatacept (Orencia), a soluble CTLA4-Ig that blocks the interaction between a co-stimulatory molecule CD28 on T cells with CD86 expressed by antigen-presenting cells [15]. Targeting of CD4 T cells has met with less success [16], however, targeting of IL-17 with an anti-IL-17 antibody may be useful for RA and potentially other autoimmune diseases [17,18]. Anti-B cell therapies for RA and SLE includeanti-CD20 [Rituximab (Rituxan)] or anti-BLyS [Belimumab (Benlysta)], which are out of the scope of the current review.
The advent of biologic agents has led to dramatic changes in the outcomes of therapies for patients with RA and other rheumatic diseases. Specific cell and cytokine targeted therapies have been successfully interrupted the inflammation mediators, disease progression, and led to the impressive advances in our understanding of the immune pathogenesis contributing to the diseases. Inflammatory macrophages and pathogenic Th17 are two critical players in autoimmune diseases. The ultimate goal of effective and safe therapy is to selectively target the central causes of the diseases and reestablish the cellular and functional homeostasis of the immune system and maintain the maximal remission. Therefore, it is conceivable that interference with the vicious cycle between inflammatory macrophages and Th17 may benefit a subset of patients that exhibit inadequate responses to the single target therapy, and might provide an important addition to the current therapeutic paradigms for RA and other rheumatic diseases [5].
Increased death receptor 5 (DR5) expression on inflammatory macrophage and T cell subpopulations: studies of the viable motheaten mice
The recessive motheaten (me/me) mouse or the allelic viable motheaten (mev/mev) mouse exhibits mutation(s) of the hematopoietic protein tyrosine phosphatase Src homology region 2 (SHP-2) domain containing phosphatase 1 (SHP-1, PTPN6) [19-21] These mice develop systemic autoimmunity including inflammatory arthritis, splenomegaly, auto antibodies, and renal diseases [22]. The predominant inflammatory cell defect in both motheaten and viable motheaten mice is accumulation of inflammatory macrophages. These macrophages exhibited increased proliferative response to GM-CSF but not to M-CSF [23]. We recently showed that the macrophages from the spleen and lymph nodes of mev/mev mice are predominately IRF-5+ and produce high levels of inflammatory cytokines including IL-23, TNF-α, IL-6 and IFN-γ [5].
Another important feature of the mev/mev mouse is that DR5, a molecule mediating apoptosis upon binding to its ligand, TRAIL, is up regulated in inflammatory macrophages. However, the DR5hi macrophages are not susceptible to apoptosis in vivo [24,25]. In further support of the pathogenic nature of these DR5hi macrophages is that these IRF-5+ macrophages also produce the highest levels of intracellular IL-23 and TNF-α [5]. Therefore, it is likely that IRF-5+ and IL-23+ M1 macrophages in the mev/mevmouse develop as a result of high levels of GM-CSF, and in part due to an apoptosis defect.
To determine whether depletion of pathogenic macrophages in mev/mev mice can be achieved by administration of TRA-8, an antihuman DR agonistic antibody, through a novel apoptosis inducing mechanism [26], and whether this can attenuate the inflammation and diseases, we crossed a novel human/mouse (hu/mo) DR5 transgenic (Tg) chimeric mouse with the mev/mev mouse which develops severe autoimmune diseases and dies prematurely [27]. This Tg mouse expresses the extracellular domain of human DR5 and the intracellular domain of mouse DR5, thereby enabling signaling through the death receptor in a mouse cells after cross linking withTRA-8, a highly-effective anti-human DR5 antibody. Treatment of these DR5 Tg+mev/mev mice resulted in significant depletion of the IRF-5+, IL-23+, and TNF-α+ subpopulation of macrophages, leading to an increased lifespan of the mice and a significant decrease of Inflammation in lung, kidney, and joints of the mice [5].
A significant population of CD4 T cells in the mev/mev mouse produced GM-CSF, and the GM-CSF subpopulation of T cells also expressed both IL-17 and IFN-γ, as well as highest levels of Tg DR5 within CD4 T cells [5]. TRA-8 treatment led to a dramatic depletion of Th1/17 (IL-17+IFN-γ+) and Th17 (IL17+IFN-γ−), but less depletion of Th1 (IFN-γ+ IL17−) cells [5].
These results indicated that both of the two cell components in the positive feedback loop, inflammatory macrophages and IL-17+ GM-CSF+ CD 4 T cells, exhibited the highest expression of Tg chimeric DR5, and administration of TRA-8 can overcome the TRAIL apoptosis defect, leading to the depletion of these two highly pathogenic cell populations.
Targeted depletion of M1 inflammatory macrophages down regulates IL-17+IFN-γ+GM-CSF+ CD4 T cells in collageninduced arthritis (CIA)
To further demonstrate that targeted depletion of M1 inflammatory macrophages can indirectly lead to the decrease of Th1/17 cells, the other component of the circuit, the Floxed STOP-hu/mo DR5 Tg mouse was crossed to LysM.Cre mice to enable the Tg chimeric DR5 in macrophages exclusively [27]. The effect of DR5 was analyzed in these LysM. Cre DR5 Tg mice in the context of CIA. TRA-8 treatment resulted in depletion of CD11b+Ly6c+ inflammatory macrophages and significantly reduced development and severity of arthritis. Histologic analysis revealed the reduction of Mac-3+ macrophages with increased apoptosis indicated by TUNEL staining and caspase live imaging in the joints. Cathepsin activity in the joints was also reduced significantly indicated by the ProSense 750 near-infrared fluorescent (NIRF) probes in anti-DR5 treated mice. Histologic examination of joints revealed a significant decrease in osteoclast activity by TRAP staining. To determine if depletion of the inflammatory M1 macrophages has an impact on the CD4 T cell population, FACS analysis of draining lymph node cells revealed that anti-hDR5 treatment depleted Th17 cells, but increased Foxp3+ T regulatory cells, which were also confirmed by real-time PCR analysis in the synovial tissues [27]. These results indicate that selective depletion of M1 macrophage by TRA-8 in a CIA mouse model can indirectly inhibit the development of inflammatory Th17 T cells, and also repopulate synovium and lymph node with Foxp3+ T regulatory cells. These results thus support a model that polarization or maintenance of inflammatory T cells requires M1 inflammatory macrophages and that a cell-based therapy of inflammatory macrophage elimination is an effective strategy to abrogate the inflammatory circuit.
Targeted depletion of Th17 T cells in RA and SLE
We have demonstrated that anti-DR5 can directly induced apoptosis in inflammatory macrophages and indirectly lead to the decrease of Th17 cells in the LysM.Cre DR5 Tg mice with CIA. However, in the mev/mev Tg DR5 mice described above, the TgDR5 expression is on both M1 macrophages and T cells. Therefore, the depletion effect of TRA-8 in Th1/17 and Th17 can be potentially direct or indirect. To clarify this, CD4+ T cells were sorted from human RA synovial fluid followed by TRA-8 treatment, which significantly reduced the Th1/17 and Th17 without the presence of M1 macrophages. This result was consistent to the high expression of DR5 in Th1/17 and Th17. Furthermore, it highly indicated that TRA-8 can directly target on pathogenic CD4 T cells in RA [5].
We previously showed that IL-17 producing T cells play an essential role in promoting spontaneous germinal center (GC) development in autoantibody production in the BXD2 mouse model of lupus [28-31]. In this mouse model, development of autoantibody producing GCs was greatly decreased by either blocking of IL-17 with a soluble IL-17R-Fc, or by ablation of IL-17R signaling in B cells using a BXD2-Il17ra-/- mouse [29]. In lupus, the one key pathogenic mechanism is the development of GCs that are essential for development of autoantibody producing B cells which are then pathogenic. The effects of anti-DR5 in blocking pathogenic auto antibodies were evaluated in the hu/mo DR5 Tg Ubc.Cre mev/mev mice. In these mice, although the transgene was mainly expressed by inflammatory macrophages and CD4 T cells but not B cells, treatment of anti-hDR5 resulted in a significant decrease in anti-DNA and antihistone IgM, IgG, IgG1, and IgG2b isotypes and led to a significantly decrease in glomerulonephritis and immune complex deposition of both IgG and IgM “isotypes” “mice”. These results together suggest the potential to prevent auto-reactive B cells via modulation of the components in the inflammatory CD4-M1 pathogenic circuit. Consistent with the results we found in RA subjects [5] and previous report [32], we have analyzed peripheral blood mononuclear cells (PBMCs) from patients with SLE and have identified that treatment with TRA-8 resulted in elimination of CD4 T cells, but not CD19 B cells (Hsu et al, unpublished observations). These results suggested that consistent with results obtained from mice and human RA, CD4 T cells, but not B cells from SLE patients could be depleted by TRA-8 therapy.
Analysis of the M1 macrophage Th17 T cell proinflammatory circuit in human RA
M1 inflammatory macrophages that produce TNF-α, IL-6 and IL-23 as well as Th17, Th1 have been proposed as key inflammatory players in RA. However, an interactive network between these macrophages and CD4 T cells has been difficult to identify using cells isolated from local inflammatory sites. We first analyzed the gene expression in the whole synovial tissues of RA subjects compared to OA subjects [5]. In RA subjects, there is a significant increase of IL-23, IRF-5 and CSF2RA (GM-CSF receptor) compared to OA, consistent with the increased abundance of M1 macrophages that produce IL-23. In the same synovial tissues, there are also increased levels of IL-17A and CSF2 (GM-CSF) suggesting that an interactive circuit between the M1 macrophages and Th17 T cells that produce GM-CSF could be operative to promote RA inflammation.
In addition to the feedback loop that is mediated by cytokines as described in Figure 1, macrophages-T cells communication can also be fulfilled by direct cell-cell contact. To demonstrate this, fresh synovial fragments were isolated from synovial fluid of RA and OA human subjects. Macrophages and T cells were then visualized by anti-CD68 and anti-CD90 staining (anti-CD90 also recognize human fibroblasts) and analyzed in confocal microscope. As shown in Figure 2A (2 dimensions) and Figure 2C (3 dimensions), the macrophage-T cell interaction is more frequent in RA compared to those in OA (Figure 2B). This observation highly suggested that there is a direct cell-cell contact between macrophages and T cells in RA synovium.
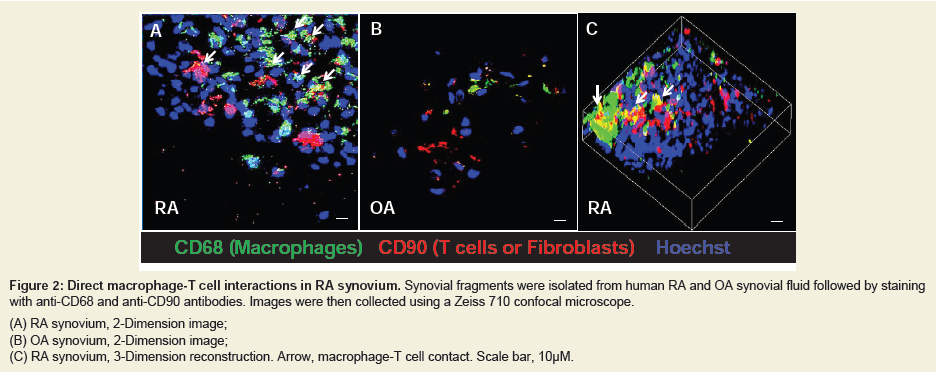
Figure 2: Direct macrophage-T cell interactions in RA synovium. Synovial fragments were isolated from human RA and OA synovial fluid followed by staining with anti-CD68 and anti-CD90 antibodies. Images were then collected using a Zeiss 710 confocal microscope.(A) RA synovium, 2-Dimension image;(B) OA synovium, 2-Dimension image;(C) RA synovium, 3-Dimension reconstruction. Arrow, macrophage-T cell contact. Scale bar, 10μM.
Summary and Conclusions
Autoimmune diseases including RA and SLE exhibit dysregulation and over activation of macrophages, T cells, as well as B cells. Major improvements have been made in the last three decades to target specific mediators of diseases and resulted in significantly improved outcomes in patients. However, certain challenges might still remain prior to the complete revealing of pathogenesis of the diseases. One of the unresolved puzzles is the hierarchy of the pathogenic events in the course of disease onset and progression. Therefore, selectively target two such interacting events, inflammatory macrophages and Th17 cells, simultaneously might exhibit higher efficacy compared to single target, especially when the hierarchy of these two components is not defined within the complexity of immune network.In this review, we have provided evidence that inflammatory IRF-5+ and IL-23+ M1 macrophages can potentiate CD4 T cells that produce IL-17 and GM-CSF. Furthermore, these two cell types form an interactive circuit and interruptions of both inflammatory cells simultaneously may provide a more potent therapeutic approach. In both RA and SLE, anti-human DR5 can be used as a model therapy to test this approach. The results show that DR5 primarily targets both the inflammatory M1 macrophages as well as pathogenic CD4 T cells. Furthermore, depletion of M1 macrophages alone indeed can, to some extent, ameliorate the Th17 T cell response, re-enforcing the concept that such a positive feedback circuit does exist. Simultaneously targeting both M1 inflammatory macrophage and Th17 pathogenic CD4 T cells is apotent therapeutic strategy for autoimmune diseases.
Acknowledgements
This work was supported by the Arthritis Foundation (to J L), the Rheumatology Research Foundation, Daiichi Sankyo Co., Ltd (to J.D.M.), and Lupus Research Institute (to H-C. H.). Additional support was granted from the Department of Veterans Affairs Merit Review Grant 1I01BX000600-01, the National Institutes of Health Grants 1AI 071110-01A1 and P30 AR048311 (to J.D.M.), and 1RO1 AI083705 (to H-C.H.).References
- Hamilton JA, Tak PP (2009) The dynamics of macrophage lineage populations in inflammatory and autoimmune diseases. Arthritis Rheum 60: 1210-1221.
- Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD (2007) Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 178: 5245-5252.
- Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, et al. (2011) IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol 12: 231-238.
- Lawrence T, Natoli G (2011) Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol 11: 750-761.
- Li J, Yang P, Wu Q, Li H, Ding Y, et al. (2013) Death Receptor 5-Targeted Depletion of Interleukin-23-Producing Macrophages, Th17, and Th1/17 Associated With Defective Tyrosine Phosphatase in Mice and Patients With Rheumatoid Arthritis. Arthritis Rheum 65: 2594-2605.
- Axtell RC, Xu L, Barnum SR, Raman C (2006) CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J Immunol 177: 8542-8549.
- Kebir H, Ifergan I, Alvarez JI, Bernard M, Poirier J, et al. (2009) Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol 66: 390-402.
- Boissier MC, Assier E, Biton J, Denys A, Falgarone G, et al. (2009) Regulatory T cells (Treg) in rheumatoid arthritis. Joint Bone Spine 76: 10-14.
- McGeachy MJ (2011) GM-CSF: the secret weapon in the T(H)17 arsenal. Nat Immunol 12: 521-522.
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A,et al. (2011) RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560-567.
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, et al. (2011) The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 12: 568-575.
- Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, et al. (2010) Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum 62: 383-391.
- Orme J, Mohan C (2012) Macrophage subpopulations in systemic lupus erythematosus. Discov Med 13: 151-158.
- Furst DE, Keystone EC, So AK, Braun J, Breedveld FC, et al. (2013) Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2012. Ann Rheum Dis 72: 2-34.
- Moreland L, Bate G, Kirkpatrick P (2006) Abatacept. Nat Rev Drug Discov 5: 185-186.
- Jendro MC, Ganten T, Matteson EL, Weyand CM, Goronzy JJ (1995) Emergence of oligoclonal T cell populations following therapeutic T cell depletion in rheumatoid arthritis. Arthritis Rheum 38: 1242-1251.
- Kellner H (2013) Targeting interleukin-17 in patients with active rheumatoid arthritis: rationale and clinical potential. Ther Adv Musculoskelet Dis 5: 141-152.
- Garber K (2012) Anti-IL-17 mAbs herald new options in psoriasis. Nat Biotechnol 30: 475-477.
- Green MC, Shultz LD (1975) Motheaten, an immunodeficient mutant of the mouse. I. Genetics and pathology. J Hered 66: 250-258.
- Shultz LD, Coman DR, Bailey CL, Beamer WG, Sidman CL (1984) "Viable motheaten," a new allele at the motheaten locus. I. Pathology. Am J Pathol 116: 179-192.
- Tsui HW, Siminovitch KA, de Souza L, Tsui FW (1993) Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet 4: 124-129.
- Su X, Zhou T, Yang P, Edwards CK 3rd, Mountz JD (1998) Reduction of arthritis and pneumonitis in motheaten mice by soluble tumor necrosis factor receptor. Arthritis Rheum 41: 139-149.
- Jiao H, Yang W, Berrada K, Tabrizi M, Shultz L, et al. (1997) Macrophages from motheaten and viable motheaten mutant mice show increased proliferative responses to GM-CSF: detection of potential HCP substrates in GM-CSF signal transduction. Exp Hematol 25: 592-600.
- Hsu HC, Shultz LD, Su X, Shi J, Yang PA, et al. (2001) Mutation of the hematopoietic cell phosphatase (Hcph) gene is associated with resistance to gamma-irradiation-induced apoptosis in Src homology protein tyrosine phosphatase (SHP)-1-deficient "motheaten" mutant mice. J Immunol 166: 772-780.
- Su X, Zhou T, Yang PA, Wang Z, Mountz JD (1996) Hematopoietic cell protein-tyrosine phosphatase-deficient motheaten mice exhibit T cell apoptosis defect. J Immunol 156: 4198-4208.
- Ichikawa K, Liu W, Fleck M, Zhang H, Zhao L, et al. (2003) TRAIL-R2 (DR5) mediates apoptosis of synovial fibroblasts in rheumatoid arthritis. J Immunol 171: 1061-1069.
- Li J, Hsu HC, Yang P, Wu Q, Li H, et al. (2012) Treatment of arthritis by macrophage depletion and immunomodulation: testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis Rheum 64: 1098-1109.
- Ding Y, Li J, Wu Q, Yang P, Luo B, et al. (2013) IL-17RA is essential for optimal localization of follicular Th cells in the germinal center light zone to promote autoantibody-producing B cells. J Immunol 191: 1614-1624.
- Hsu HC, Yang P, Wang J, Wu Q, Myers R, et al. (2008) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9: 166-175.
- Wang JH, New JS, Xie S, Yang P, Wu Q, et al. (2013) Extension of the Germinal Center Stage of B Cell Development Promotes Autoantibodies in BXD2 Mice. Arthritis Rheum 65: 2703-2712.
- Xie S, Li J, Wang JH, Wu Q, Yang P, et al. (2010) IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol 184: 2289-2296.
- Crowder RN, Zhao H, Chatham WW, Zhou T, Carter RH (2011) B lymphocytes are resistant to death receptor 5-induced apoptosis. Clin Immunol 139: 21-31.