
Journal of Parkinsons disease and Alzheimers disease
Download PDF
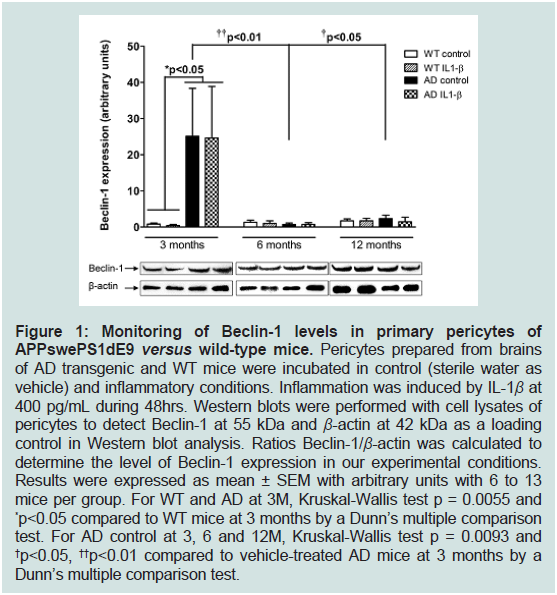 Figure 1: Monitoring of Beclin-1 levels in primary pericytes of
APPswePS1dE9 versus wild-type mice. Pericytes prepared from brains
of AD transgenic and WT mice were incubated in control (sterile water as
vehicle) and inflammatory conditions. Inflammation was induced by IL-1β at
400 pg/mL during 48hrs. Western blots were performed with cell lysates of
pericytes to detect Beclin-1 at 55 kDa and β-actin at 42 kDa as a loading
control in Western blot analysis. Ratios Beclin-1/β-actin was calculated to
determine the level of Beclin-1 expression in our experimental conditions.
Results were expressed as mean ± SEM with arbitrary units with 6 to 13
mice per group. For WT and AD at 3M, Kruskal-Wallis test p = 0.0055 and
*p<0.05 compared to WT mice at 3 months by a Dunn’s multiple comparison
test. For AD control at 3, 6 and 12M, Kruskal-Wallis test p = 0.0093 and
†p<0.05, ††p<0.01 compared to vehicle-treated AD mice at 3 months by a
Dunn’s multiple comparison test.
Figure 1: Monitoring of Beclin-1 levels in primary pericytes of
APPswePS1dE9 versus wild-type mice. Pericytes prepared from brains
of AD transgenic and WT mice were incubated in control (sterile water as
vehicle) and inflammatory conditions. Inflammation was induced by IL-1β at
400 pg/mL during 48hrs. Western blots were performed with cell lysates of
pericytes to detect Beclin-1 at 55 kDa and β-actin at 42 kDa as a loading
control in Western blot analysis. Ratios Beclin-1/β-actin was calculated to
determine the level of Beclin-1 expression in our experimental conditions.
Results were expressed as mean ± SEM with arbitrary units with 6 to 13
mice per group. For WT and AD at 3M, Kruskal-Wallis test p = 0.0055 and
*p<0.05 compared to WT mice at 3 months by a Dunn’s multiple comparison
test. For AD control at 3, 6 and 12M, Kruskal-Wallis test p = 0.0093 and
†p<0.05, ††p<0.01 compared to vehicle-treated AD mice at 3 months by a
Dunn’s multiple comparison test.
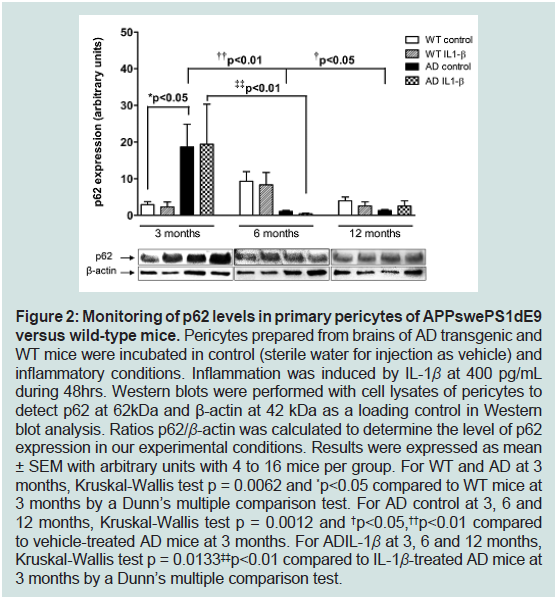 Figure 2: Monitoring of p62 levels in primary pericytes of APPswePS1dE9
versus wild-type mice. Pericytes prepared from brains of AD transgenic and
WT mice were incubated in control (sterile water for injection as vehicle) and
inflammatory conditions. Inflammation was induced by IL-1β at 400 pg/mL
during 48hrs. Western blots were performed with cell lysates of pericytes to
detect p62 at 62kDa and β-actin at 42 kDa as a loading control in Western
blot analysis. Ratios p62/β-actin was calculated to determine the level of p62
expression in our experimental conditions. Results were expressed as mean
± SEM with arbitrary units with 4 to 16 mice per group. For WT and AD at 3
months, Kruskal-Wallis test p = 0.0062 and *p<0.05 compared to WT mice at
3 months by a Dunn’s multiple comparison test. For AD control at 3, 6 and
12 months, Kruskal-Wallis test p = 0.0012 and †p<0.05,††p<0.01 compared
to vehicle-treated AD mice at 3 months. For ADIL-1β at 3, 6 and 12 months,
Kruskal-Wallis test p = 0.0133‡‡p<0.01 compared to IL-1β-treated AD mice at
3 months by a Dunn’s multiple comparison test.
Figure 2: Monitoring of p62 levels in primary pericytes of APPswePS1dE9
versus wild-type mice. Pericytes prepared from brains of AD transgenic and
WT mice were incubated in control (sterile water for injection as vehicle) and
inflammatory conditions. Inflammation was induced by IL-1β at 400 pg/mL
during 48hrs. Western blots were performed with cell lysates of pericytes to
detect p62 at 62kDa and β-actin at 42 kDa as a loading control in Western
blot analysis. Ratios p62/β-actin was calculated to determine the level of p62
expression in our experimental conditions. Results were expressed as mean
± SEM with arbitrary units with 4 to 16 mice per group. For WT and AD at 3
months, Kruskal-Wallis test p = 0.0062 and *p<0.05 compared to WT mice at
3 months by a Dunn’s multiple comparison test. For AD control at 3, 6 and
12 months, Kruskal-Wallis test p = 0.0012 and †p<0.05,††p<0.01 compared
to vehicle-treated AD mice at 3 months. For ADIL-1β at 3, 6 and 12 months,
Kruskal-Wallis test p = 0.0133‡‡p<0.01 compared to IL-1β-treated AD mice at
3 months by a Dunn’s multiple comparison test.
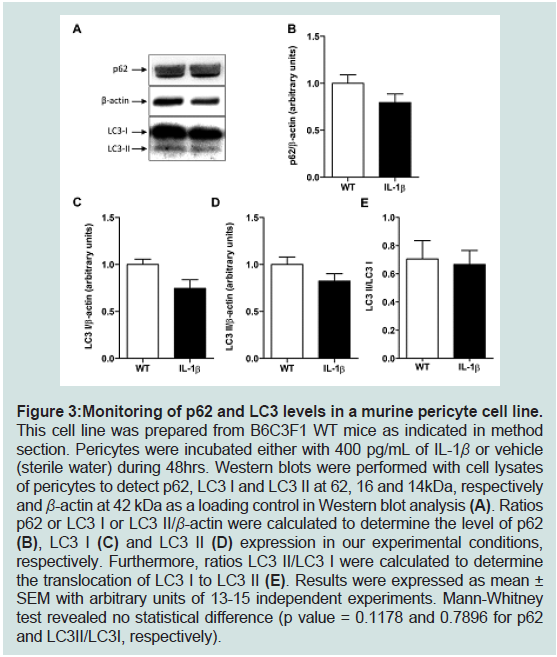 Figure 3: Monitoring of p62 and LC3 levels in a murine pericyte cell line. This cell line was prepared from B6C3F1 WT mice as indicated in method
section. Pericytes were incubated either with 400 pg/mL of IL-1β or vehicle
(sterile water) during 48hrs. Western blots were performed with cell lysates
of pericytes to detect p62, LC3 I and LC3 II at 62, 16 and 14kDa, respectively
and β-actin at 42 kDa as a loading control in Western blot analysis (A). Ratios
p62 or LC3 I or LC3 II/β-actin were calculated to determine the level of p62
(B), LC3 I (C) and LC3 II (D) expression in our experimental conditions,
respectively. Furthermore, ratios LC3 II/LC3 I were calculated to determine
the translocation of LC3 I to LC3 II (E). Results were expressed as mean ±
SEM with arbitrary units of 13-15 independent experiments. Mann-Whitney
test revealed no statistical difference (p value = 0.1178 and 0.7896 for p62
and LC3II/LC3I, respectively).
Figure 3: Monitoring of p62 and LC3 levels in a murine pericyte cell line. This cell line was prepared from B6C3F1 WT mice as indicated in method
section. Pericytes were incubated either with 400 pg/mL of IL-1β or vehicle
(sterile water) during 48hrs. Western blots were performed with cell lysates
of pericytes to detect p62, LC3 I and LC3 II at 62, 16 and 14kDa, respectively
and β-actin at 42 kDa as a loading control in Western blot analysis (A). Ratios
p62 or LC3 I or LC3 II/β-actin were calculated to determine the level of p62
(B), LC3 I (C) and LC3 II (D) expression in our experimental conditions,
respectively. Furthermore, ratios LC3 II/LC3 I were calculated to determine
the translocation of LC3 I to LC3 II (E). Results were expressed as mean ±
SEM with arbitrary units of 13-15 independent experiments. Mann-Whitney
test revealed no statistical difference (p value = 0.1178 and 0.7896 for p62
and LC3II/LC3I, respectively).
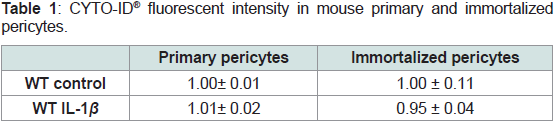 Table 1: CYTO-ID® fluorescent intensity in mouse primary and immortalized
pericytes.
Pericytes were exposed to 400 pg/mL IL-1β for 48hours and CYTO-ID® dual
color reagent was added for 30 min at room temperature before being read by
fluorescence using a Varioskan Flash microplate reader. Rapamycin (1 μM)
as an inducer of autophagy and Chloroquine (10 μM) as a lysosomal inhibitor
were included as positive controls. Ratios of fluorescent intensity for CYTO-ID®
green detection reagent / fluorescent intensity for Hoechst 33342 nuclear stain
were calculated and results were normalized to control. For Rapamycin and
Chloroquine mixture, ratio was 1.32 ± 0.04.
Table 1: CYTO-ID® fluorescent intensity in mouse primary and immortalized
pericytes.
Pericytes were exposed to 400 pg/mL IL-1β for 48hours and CYTO-ID® dual
color reagent was added for 30 min at room temperature before being read by
fluorescence using a Varioskan Flash microplate reader. Rapamycin (1 μM)
as an inducer of autophagy and Chloroquine (10 μM) as a lysosomal inhibitor
were included as positive controls. Ratios of fluorescent intensity for CYTO-ID®
green detection reagent / fluorescent intensity for Hoechst 33342 nuclear stain
were calculated and results were normalized to control. For Rapamycin and
Chloroquine mixture, ratio was 1.32 ± 0.04.
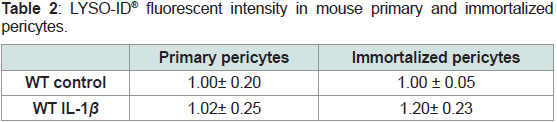 Table 2: LYSO-ID® fluorescent intensity in mouse primary and immortalized
pericytes.
Pericytes were exposed to 400 pg/mL IL-1β for 48 hours and LYSO-ID® dual
color reagent was added for1 hour at room temperature before being read by
fluorescence using a Varioskan Flash microplate reader. verapamil (10 μM),
a lysosome-perturbation agent, is provided as a positivecontrol. Ratios of
fluorescent intensity for LYSO-ID® red dye signal /fluorescent intensity for the
blue nuclear counterstain were calculated and results were normalized to control.
For Verapamil, ratio was 1.32 ± 0.12.
Table 2: LYSO-ID® fluorescent intensity in mouse primary and immortalized
pericytes.
Pericytes were exposed to 400 pg/mL IL-1β for 48 hours and LYSO-ID® dual
color reagent was added for1 hour at room temperature before being read by
fluorescence using a Varioskan Flash microplate reader. verapamil (10 μM),
a lysosome-perturbation agent, is provided as a positivecontrol. Ratios of
fluorescent intensity for LYSO-ID® red dye signal /fluorescent intensity for the
blue nuclear counterstain were calculated and results were normalized to control.
For Verapamil, ratio was 1.32 ± 0.12.
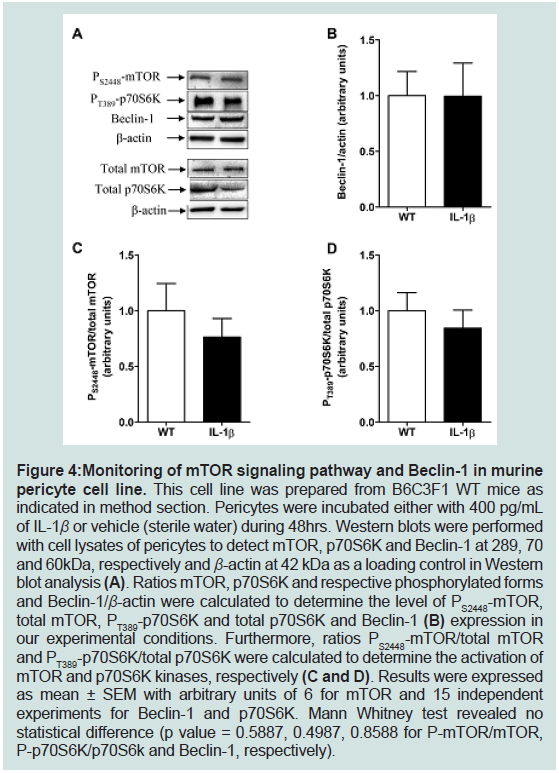 Figure 4: Monitoring of mTOR signaling pathway and Beclin-1 in murine
pericyte cell line. This cell line was prepared from B6C3F1 WT mice as
indicated in method section. Pericytes were incubated either with 400 pg/mL
of IL-1β or vehicle (sterile water) during 48hrs. Western blots were performed
with cell lysates of pericytes to detect mTOR, p70S6K and Beclin-1 at 289, 70
and 60kDa, respectively and β-actin at 42 kDa as a loading control in Western
blot analysis (A). Ratios mTOR, p70S6K and respective phosphorylated forms
and Beclin-1/β-actin were calculated to determine the level of PS2448-mTOR,
total mTOR, PT389-p70S6K and total p70S6K and Beclin-1 (B) expression in
our experimental conditions. Furthermore, ratios PS2448-mTOR/total mTOR
and PT389-p70S6K/total p70S6K were calculated to determine the activation of
mTOR and p70S6K kinases, respectively (C and D). Results were expressed
as mean ± SEM with arbitrary units of 6 for mTOR and 15 independent
experiments for Beclin-1 and p70S6K. Mann Whitney test revealed no
statistical difference (p value = 0.5887, 0.4987, 0.8588 for P-mTOR/mTOR,
P-p70S6K/p70S6k and Beclin-1, respectively).
Figure 4: Monitoring of mTOR signaling pathway and Beclin-1 in murine
pericyte cell line. This cell line was prepared from B6C3F1 WT mice as
indicated in method section. Pericytes were incubated either with 400 pg/mL
of IL-1β or vehicle (sterile water) during 48hrs. Western blots were performed
with cell lysates of pericytes to detect mTOR, p70S6K and Beclin-1 at 289, 70
and 60kDa, respectively and β-actin at 42 kDa as a loading control in Western
blot analysis (A). Ratios mTOR, p70S6K and respective phosphorylated forms
and Beclin-1/β-actin were calculated to determine the level of PS2448-mTOR,
total mTOR, PT389-p70S6K and total p70S6K and Beclin-1 (B) expression in
our experimental conditions. Furthermore, ratios PS2448-mTOR/total mTOR
and PT389-p70S6K/total p70S6K were calculated to determine the activation of
mTOR and p70S6K kinases, respectively (C and D). Results were expressed
as mean ± SEM with arbitrary units of 6 for mTOR and 15 independent
experiments for Beclin-1 and p70S6K. Mann Whitney test revealed no
statistical difference (p value = 0.5887, 0.4987, 0.8588 for P-mTOR/mTOR,
P-p70S6K/p70S6k and Beclin-1, respectively).
Research Article
Autophagy Monitoring in Cerebral Pericytes from Alzheimer’s disease Mouse Model in an Inflammatory Environment
Julie V, Vincent T, Hanitriniaina R†, Benjamin F, Thierry F and Guylène P*
University of Poitiers, Neurovascular Unit and Cognitive Disorders,
PôleBiologieSanté, Poitiers, France
†Present address: University of Poitiers, EA4331, Laboratoire
Inflammation, Tissus Epithéliaux et Cytokines, Pôle Biologie Santé,
Poitiers, France
*Address for Correspondence: Guylène P, University of Poitiers, Neurovascular Unit and Cognitive
Disorders, PôleBiologieSanté, Poitiers, France; E-mail: guylene.page@
univ-poitiers.fr
Submission: 01 August, 2022
Accepted: 27 September, 2022
Published: 03 October, 2022
Copyright: © 2022 Ben-Julie V, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background:
The blood-brain barrier (BBB) is a complex neurovascular
unit involving pericytes as multi-functional cells that play a crucial role in
maintaining homeostasis. In Alzheimer’s disease (AD), platelet-derived
growth factor receptor-β (PDGFRβ) immunostaining revealed significantly
reduced pericyte coverage of brain capillaries as well as reduced pericyte
numbers in AD cortex and hippocampus compared with control brains.
However, the mechanisms of pericyte loss have yet to be completely defined.
Moreover, we have previously shown that, in microglia, interleukin-1β (IL-
1β)-induced inflammation blocks autophagic flow, a physiological process
involved in the degradation of proteins including the β-amyloid peptide. Thus,
we evaluated whether the inflammatory response in AD impaired autophagy
in pericytes.
Methods:
A longitudinal autophagic status monitoring was performed
in pericytes purified from brains of AD and wild type (WT) mice at 3, 6 and
12 months. Furthermore, the impact of an inflammatory environment was
studied not only in these primary pericytes but also in a pericyte cell line
developed in the laboratory.
Results:
Primary pericytes from AD mice displayed a significant increase
of autophagic markers at 3 months and that in later stages their expressions
were like those of WT mice. In addition, IL-1β-induced inflammation did not
modify the expression of autophagic markers or those of mTOR signaling
pathway in both primary and immortalized mouse pericytes.
Conclusions:
For the first time, these data highlighted that autophagy
is activated in primary pericytes from AD transgenic mice at 3 months.
In addition, inflammation has no impact on autophagic flow under our
experimental conditions.
Keywords
Pericytes; Alzheimer’s disease; Inflammation, Autophagy,
Interleukin-1β
Introduction
We have previously published the first results on the interaction
between autophagy and inflammation in Alzheimer’s disease (AD)
[1-3]. We showed that the pro-inflammatory cytokine interleukin-
1β (IL-1β) was responsible for blocking autophagy and that microglia
was the most sensitive cell type in primary neuron/astrocyte/
microglia tri-cultures [1]. These last results are consistent with recent
data concerning the failure of these brain resident immune cells
during AD [4]. However, other cells showed senescence and died in
AD such as pericytes [5]. Recently, authors showed elevated soluble
platelet-derived growth factor receptor-β (sPDGFRβ) levels in
cerebrospinal fluid (CSF), indicating pericyte injury and blood brain
barrier (BBB) breakdown [6,7]. So, sPDGFRβ could be a promising
early biomarker of human cognitive dysfunction [7,8]. Pericytes are
mural cells abundant in the microvasculature in the central nervous
system and physically the closest cells to brain endothelial cells (EC)
wrapping around them, joined by gap junctions, and interfacing
bypeg-and-socket structures [9,10]. Their functions are largely explored from embryonic to adult periods of development [6,11-13].
Under physiological conditions, pericytes regulate BBB integrity,
angiogenesis, phagocytosis, cerebral blood flow (CBF) and capillary
diameter, neuro inflammation and multi potent stem cell activity [9-14].The presence of pericytes is essential because knock-out mouse
models by deletion and/or genetic manipulation of PDGFRβ and/
or PDGFB genes result in blood vessel dilation, EC hyperplasia, and
micro aneurysm formation [15]. Furthermore, in double transgenic
APPsw/0/PDGFRβ+/- mice, the loss of pericytes early increased brain
β-amyloid peptide (Aβ) levels, accelerated amyloid angiopathy and
cerebral β-amyloidosis, led to the development of tau pathology and
an early neuronal loss that is normally absent in APP transgenic mice
[16,17]. Many studies showed that pericyte dysfunction is associated
with AD neuropathology [18]. In fact, pericyte degeneration led to
BBB disruption and unrestricted entry and accumulation of bloodderived
products in brain (erythrocyte-derived hemoglobin and
plasma-derived proteins such as albumin, plasmin, thrombin, fibrin,
immunoglobulins and others) [19]. In humans with cognitive decline,
A β constricted brain capillaries at pericyte locations and generated
reactive oxidative species (ROS) which led to release of endothelin-1
acting via endothelin receptors (ETA) on pericytes and increasing
the constricting effects [20]. Neuro imaging studies in individuals
with mild cognitive impairment (MCI) and early AD revealed BBB
breakdown in the hippocampus before brain atrophy and dementia
[21-23]. The mechanisms of pericyte loss have yet to be completely
defined. AD is also characterized by a great inflammatory response and
by a blockage of autophagy [24-29]. In AD, both neuro inflammation
and systemic inflammation can disrupt the BBB by modifying the
tight junctions, damaging the vascular endothelial cell, degrading
glycocalyx, allowing peripheral hematopoietic cells to infiltrate neural
tissue and become part of the parenchymal microglia/macrophage
[23,24,30]. Notably, IL-1β contributes to BBB dysfunction [25,31].
Pericytes express several mediators (chemokines, cytokines) that
can enhance leukocyte extravasation and contribute to a cerebral
inflammatory phenotype [32,33]. At the same time, pericytes over express adhesion molecules that guide and instruct innate immune
cells after transendothelial migration [32]. Moreover, pericytes are
implicated in shaping adaptive immunity, with several studies that
point to an immunosuppressive role [34]. However, in AD, pericyte
loss also correlates with inflammation-mediated disruption of BBB
[32]. It is well known that inflammatory response in particular IL-
1β production impairs autophagy resulting in neuro degeneration
and memory loss in AD [1-3,35]. However, the autophagic status of
pericytes in AD is still poorly understood. Therefore, this study aims
to explore the autophagic status of pericytes from AD mice versus
wild type (WT) mice at 3, 6 and 12 months. The analysis focused on
the expression of Beclin-1, a protein involved in autophagic initiation
[36], p62 as a cargo protein of the material to be eliminated [37],
and soluble LC3 I and membranaryunsoluble LC3 II isoforms as
elongation and fusion signals with lysosomes [38]. Furthermore, a
pericyte cell line developed in the laboratory was also used to study
the impact of IL-1β in autophagy and mTOR signaling pathway.
Materils & Methods
Chemical products:
Sodium fluoride (NaF), phenylmethylsulfonyl fluoride (PMSF),
protease and phosphatase inhibitor cocktails, dithiothreitol (DTT),
mouse monoclonal Anti-β-Actin antibody, mouse anti-alpha smooth
muscle actin antibody (αSMA), and all reagent-grade chemicals for
buffers were purchased from Sigma (St Quentin Fallavier, France),
sodium pentobarbital from CEVA, Animal Health (Libourne,
France), 4X Laemmli sample buffer, 4-15% mini-PROTEAN® TGX™
gels, Tris-glycine running buffer and Trans-Blot® Turbo™ Transfer
System from Biorad (Marnes-la-Coquette, France),Quant-it® protein
assay. For western blot, primary antibodies against Beclin-1, mTOR,
p70S6K (total and phosphorylated forms), mouse anti-GFAP for
immunocytofluorescence, secondary anti-rabbit or anti-mouse
IgG antibody conjugated with Horseradish Peroxydase (HRP) and
mouse recombinant Interleukin-1β (IL-1β)were purchased from Cell
Signalling (Ozyme, St Quentin Yvelines, France) except p62/SQSTM1
and LC3I/II from MBL (CliniSciences distributor, Nanterre, France),
rabbit polyclonal anti-NG2 Chondroitin Sulfate Proteoglycan (NG2)
and anti-platelet derived growth factor receptor beta (PDGFRβ)
antibodies from Proteintech France, a rabbit polyclonal antibody
anti-von Willebrand Factor (vWF) from Merck Millipore (Molsheim,
Alsace France), macrosialin or murine homologue of the human
CD68 and secondary antibodies goat anti-rat R-Phycoerythrin (RPE)
from AbDSerotec(Düsseldorf, Germany), IgG- and protease-Free
Bovine Serum Albumin (BSA) and secondary anti-mouse-Alexa 488
or anti-rabbit-TRITC from Jackson Immuno Research Europe Ltd
(Interchim distributor, Montluçon, France).
Cell culture:
For this work, mouse pericytes were extracted following
experimental protocol described in our patents (FR17/57643 and
US-2022-0010258-A1). Two cultures of mouse pericytes were used:
a mouse primary culture of pericytes and a mouse cell line of
pericytes developed in the laboratoryas indicated in the patent.
Pericytes came from brains of APPswePS1dE9 or wild type
(WT) mice at 3, 6 and 12 months of age. These transgenic mice were
purchased from Mutant Mouse Resources and Research Centers (Stock No: 34829-JAX, USA) displaying Alzheimer phenotype
(Authorization from “Haut Comité de Biotechnologiefrançais”
(HCB) to Pr Guylène Page, number 2040 for reproduction, treatment,
behavioral tests and ex-vivo experiments). Wild type (WT) with
B6C3F1 background and APPswePS1dE9 mice were obtained by
crossing a male APPswePS1dE9 mouse with a WT female mouse (from
Charles River, strain Code 031) as explained previously [39].The use
of animals was approved by the Ethical and Animal Care Committee
(N˚84 COMETHEA, Ethical Committee for Animal Experimentation
Poitou-Charentes, France). At weaning, all mice were genotyped by
polymerase chain reaction (PCR) analysis of tail biopsies according
to the manufacturer’s recommended protocols. All animal care and
experimental procedures conformed with the French Decree number
2013–118, 1 February 2013 NOR: AGRG1231951D in accordance
with European Community guidelines (directive 2010/63/UE). All
efforts were made to minimize animal suffering, as well as the number
of animals used. The animals were housed in a conventional state
under adequate temperature (23 ± 3˚C) and relative humidity (55 ±
5%) control with a 12/12 h reversed light/dark cycle with access to
food and water ad libitum.
As a method of immortalization, we used the transformation
with oncoprotein of murine polyomavirus, Polyoma middle T
antigen. Protocol was already described for murine endothelial cells
[40]. Stable cerebral pericyte cell line has been obtained 2 months
later. This pure cell line displayed features of pericytes with αSMA,
NG2, PDGFRβ and no vWF, ZO-1, GFAP nor CD68 (markers of
endothelial cells, astrocytes and microglia, respectively) expressions
as shown in additional file 1 (see Supplementary Information).
Cell treatment:
Primary pericytes and immortalized pericyteswere seeded in
24-well plates (150,000 and 50,000 cells/well, respectively) to study
their autophagic status in control or inflammatory conditions.
Inflammation was induced by IL-1β at 400 pg/mL during 48 hours
in a cell incubator. After 48hr-treatment, cells were lysed in ice-cold
lysis buffer (50 mM Tris-HCl, 50 mM NaCl pH 6.8, 1% (v/v) Triton
X-100, 1 mM PMSF, 50 mM NaF, 1% (v/v) protease inhibitor and 1%
(v/v) phosphatase inhibitor cocktails). Lysates were sonicated for 10
sec and centrifuged at 15,000 × g for 15 min at 4°C. The supernatants
were collected and analyzed for protein determination using a Quantit
® protein assay kit. Samples were frozen at -80°C until western blot
experiments.
Western blot:
Samples (40 μg proteins) were prepared for electrophoresis by
adding 4X Laemmli sample buffer containing 0.05 M DTT and loaded
into 4-15% mini-PROTEAN® TGX™ gels with Tris-glycine SDS
running buffer. Systems ran at 200 V for 35 minutes. Then, gels were
transferred to nitrocellulose membranes using Trans-Blot® Turbo™
Transfer System (25V, 3 min for 0.2 μm nitrocellulose MINI gel).
Membranes were washed for 10 min in Tris-buffered saline/Tween
(TBST: 20 mM Tris-HCl, 150 mM NaCl, pH 7.5, 0.05% Tween 20)
and aspecific antigenic sites were blocked 2h in TBST containing 10%
BSA for detection of LC3 and in TBST containing 5% non-fat milk for
other proteins. Antibodies used were rabbit anti-Beclin-1, anti-p62,
anti-LC3, anti-total mTOR, anti-total p70S6K, anti-PS2448-mTOR, anti-PT389-p70S6K all at a dilution 1:500 in TBST containing 5% BSA
overnight at 4°C. Membranes were washed twice with TBST and then
incubated with the HRP-conjugated secondary antibody anti-rabbit
IgG (1:1000), during 1 hour at RT. Membranes were washed again
and exposed to the chemiluminescence Luminata Forte Western HRP
Substrate (Millipore, Saint-Quentin-en-Yvelines, France) followed by
signal’s capture with the Gbox system (GeneSnap software, Syngene,
Ozyme distributor). After 2 washes in TBST, membranes were probed
with mouse antibody against β-actin (1:1000) overnight at 4°C.
They were then washed with TBST, incubated with HRP-conjugated
secondary antibody anti-mouse (1:1000 in blocking buffer) for 1h,
exposed to the chemiluminescence Luminata Classico Western HRP
Substrate (Millipore, Saint-Quentin-en-Yvelines, France) and signals
were captured. Automatic image analysis software is supplied with
Gene Tools (Syngene, Ozyme distributor). Protein/β-actinratios were
calculated and showed in the corresponding figures.
CYTO-ID<sup>®</sup> Autophagy Detection Kit:
This live cell analysis kit provides a convenient approach for the
analysis of the regulation of autophagic activity at the cellular level.
Cell treatment was performed in 96-well plates (1,25,000 cells/well).
According to the supplier’s recommendations, cell medium was
gently removed after treatment and cells were washed with 200 μL
of 1X assay buffer containing 5% NBCS. Then, 100μL of 1X Assay
Buffer/5% NBCS containing 2μL/mL of CYTO-ID® green detection
reagent and 2μL/mL Hoechst 33342 nuclear stain were added in
each well. Plates were incubated 30 min at room temperature in a
black chamber. Then, cells were washed twice with 100 μL of 1X
assay buffer/5% NBCS and 100 μL of this buffer were added for
measurement of fluorescent signals (λExcitation: 480nm/λEmission:
530nm for CYTO-ID® green detection reagent and λExcitation:
340nm/λEmission: 480nm for Hoechst 33342 nuclear stain) by
using a ThermoVarioskan Flash spectral scanning multimode reader
(Thermo Fisher Scientific, Illkirch, France).Rapamycin (1μM)as
an inducer of autophagy and Chloroquine (10 μM) as a lysosomal
inhibitor were included as positive controls. Ratios of fluorescent
intensity for CYTO-ID® green detection reagent /fluorescent intensity
for Hoechst 33342 nuclear stain were calculated and results were
normalized to control.
LYSO-ID<sup>®</sup> Red Cytotoxicity Kit:
This GFP certified® live cell kit provides a rapid and quantitative
approach for determining drug- or toxic agent-induced lysosome and
lysosome-like organelle perturbations, for detecting phospholipidosis
and also the accumulation of autophagosomes by blocking the
downstream lysosomal pathway and/or intracellular trafficking
of autophagosomes also lead to increase in the accumulation of
intracellular LYSO-ID® red dye signal. A lysosome-perturbation agent,
verapamil (10 μM), is provided as a positive control for monitoring
changes in vacuole number and volume. A blue nuclear counter stain
is integrated into the detection reagentto identify cell death or loss. As
indicated for CYTO-ID®, cells were seeded in 96-well plates (1,25,000
cells/well) and treated with vehicle or IL-1β. After treatment, medium
was carefully aspirated and 100μl of 1X Assay Buffer containing 2%
of NBCS were added in each well. Then, the buffer was aspirated and
100μl of the 1X dual color detection reagent containing 1mL Dual
Color Detection Reagent, 8.8 mL Detection Buffer and 2% of NBCS for 10 mL of solution was added in each well. Plates were incubated
for 1 hour at room temperature and protected from light. At the end
of incubation, plates were gently washed twice with 100 μL of 1X
Assay Buffer/2% of NBCS and 80 μL of 1X Assay Buffer/2% of NBCS
were added before measurement of intensity of fluorescence. The red
lysosome stain can be read witha λExcitation: 540nm/λEmission:680
nm and the blue nuclear counterstain can be read with a λExcitation:
340nm/λEmission: 480nm. Ratios of fluorescent intensity for
LYSO-ID® red dye signal /fluorescent intensity for the blue nuclear
counterstain were calculated and results were normalized to control.
Statistical analysis:
For biochemical analysis, results are expressed as means ±
SEM. To compare quantitative variables between primary pericytes
prepared from WT and APPswePS1dE9 mice treated or not with IL-
1β and between immortalized pericytes treated or not with IL-1β,
Mann-Withney’s tests were used. Longitudinal changes in parameters
occurring during the life were analysed with a Kruskal-Wallis test
followed by a post-hoc with Dunns test (GraphPad Instat, GraphPad
Software, San Diego, CA, USA). The level of significance was p < 0.05.
Results
Monitoring of autophagy in primary mouse pericytes:
To determine whether autophagy changes occurred in pericytes
prepared from brains of APPswePS1dE9 mice at 3, 6 and 12 months
of age, immunoblottings of Beclin-1, p62, LC3 I and LC3 II were
performed. The levels of expression of Beclin-1 which is a key
component in the initiation of autophagosome formation significantly
increased by 31-fold in pericytes of 3-months old APPswePS1dE9
mice compared to age-matched WT mice [41]. On the contrary, a
robust decrease by 36- and 10.8-fold in pericytes of APPswePS1dE9
mice was observed at 6 and 12 months, respectively compared to
3-months old APPswePS1dE9 mice (Figure 1).
The protein p62 is an autophagic receptor which recognizes
ubiquitinylated proteins and interacts with LC3 II at the forming
autophagosome [37,42]. As for Beclin-1, results showed a great
increase of its expression levels (6.8-fold) in pericytes from 3-months
old APPswePS1dE9 mice compared to age-matched WT mice,
whereasp62 levels were significantly decreased by 18.5- and 14-
fold in pericytes prepared from brains of 6 and 12 months old
APPswePS1dE9 mice, respectively versus transgenic mice at 3 months
old age (Figure 2).
Contrary to primary microglia [1], an inflammatory environment
induced by IL-1β did not influence the levels of autophagic parameters
in primary pericytes (Figure 1 and 2). To complete these results, we
performed CYTO-ID® and LYSO-ID® assays to determine if there is an
autophagosome accumulation (CYTO-ID® and LYSO-ID® responses)
or lysosome and lysosome-like organelle perturbations and
phospholipidosis (LYSO-ID® response) at 3 months. No difference
of the fluorescent intensity was observed in primary WT pericytes
exposed to 400 pg/mL IL-1β compared to vehicle-treated pericytes
(Table 1 and 2).
Monitoring of autophagy in mouse immortalized pericytes:
As indicated in the section of materials and methods, a murine
WT pericyte line was obtained from primary pericytes extracted from
the brains of 3-month-old B6C3F1 mice. This line allows us to obtain
more biological material than primary cells and to reduce the number
of animals in the experiments. It seemed useful to us to check whether
this line responded similarly in their autophagic components,
as mouse primary pericyte cultures in the same inflammatory
environment. As shown in figure3, IL-1β induced no modification of
p62 (panels A,B) norLC3 I and LC3 II (panels A, C to E) expression
levels. These results are also reinforced by the absence of changes in
Beclin-1 expression and in mTOR and p70S6K activations in IL-1β-
treated pericytes compared to control pericytes (Figure 4).
We also performed CYTO-ID® and LYSO-ID® experiments
with the cell line. The fluorescent intensities were similarwhatever
conditionto primary pericyte results (Table 1 and 2).
Discussion
Autophagy dysfunction was detected in brains of AD patients in
2005 [27]. Many authors then published data validating this alteration
with a decrease in Beclin-1 level and its role in AD [36,43,44], a
blockage of autophagic flow with dysfunction of lysosomal activities
during AD in various experimental models of the disease [28,29,45].
Previous results showed that IL-1β led to a blockage of autophagy
in microglia, highly sensitive to this inflammatory stress [1]. Other
authors demonstrated the relationship between autophagy and IL-
1β stress in AD [35]. Thus, chemical modulators of autophagy as
well as gene therapy targeting autophagy related proteins offer great
potential for the AD treatment. A number of mTOR-dependent
and independent autophagy modulators have been demonstrated to
have positive effects in AD animal models and patients [28,46,47].
However, no data are available in the literature about autophagy at
the level of BBB during aging or in AD, while the BBB dysfunction in AD is largely described [16,19,48]. Studies showed that autophagy
was enhanced in middle cerebral artery occlusion/reperfusion and
oxygen-glucose deprivation/reperfusion experiments. In these
contexts, autophagy would have either protective or detrimental
roles in BBB function and ECs [49]. Autophagy could decrease BBB
permeability and ischemic damage, but also induce apoptosis and cell
death [49].
For the first time, we observed that primary pericytes purified
from brain of APPswePS1dE9 mice showed an increase of Beclin-1,
p62 and LC3 II expression levels at 3 months compared to agematched
WT mice. Then, levels of these autophagy markers were like
those of WT mice at 6 and 12 months. These autophagy changes in
primary pericytes at 3 months revealed an activation of autophagy
with no accumulation of autophagosomes or lysosomes, indicating no
impairment of autophagic flow. At 3 months, APPswePS1dE9 mice
displayed no amyloid deposits but produced intracellular Aβ. At 6
months, extracellular Aβ was mainly depicted in cerebral parenchyma
[50]. It is known that Aβ34, a marker for amyloid clearance in
AD, was predominantly detectable in a subset of brain capillaries
associated with pericytes, while in later clinically diagnosed AD
stages, this pericyte-associated Aβ34 immuno reactivity was largely
lost [51]. BBB-associated pericytes clear Aβ aggregates via an LRP1-
dependentApoE isoform-specific mechanism with apoE4 disrupting
Aβ clearance compared to apoE3 [52,53]. One may propose that
enhanced autophagy at 3 months in these transgenic AD mice could
be explained by the proteolytic degradation of Aβ40 and Aβ42.
Contrary to primary microglia [1,35], IL-1β-induced inflammation
did not modify expression of Beclin-1, p62 and LC3 II in primary WT
or AD pericytes. Furthermore, no modification of autophagy markers
and of the mTOR signaling pathway was observed in pericyte cell
line. The concentration of IL-1β (400 pg/mL) may be questioned. It
was chosen based on our previous work on microglia where higher
concentrations led to cell death [1]. Recently, authors showed that
inhibition of the NLRP3-contained inflammasome reducedpericyte
cell coverage and decreased protein level of PDGFR β contrary to
IL-1β which is the major product of NLRP3 [54]. Concentrations
of IL-1β were 12.5 to 125 times higher than those used in our study
and authors concluded that NLRP3 activation maintained healthy
pericytes in the brain and warned of therapeutic strategies to inhibit
inflammasome [54]. Besides the relationship between NLRP3 and
pericyte survival, we can ask the question of the resistance of the
pericytes to this inflammatory stress at the autophagic level because
inflammation generally stimulates autophagy, or even blocks the flow
and leads to death. However, no data were available in the literature
about the autophagic status of pericytes in AD. In stroke and cocaine
exposure, authors showed that in pericytes autophagy was regulated
by the sigma-1 receptor signaling pathway [55,56].
For the first time, we show that autophagy is activated in primary
pericytes at 3 months. Proteolytic degradation of amyloid peptides
may explain this activation. In addition, inflammation has no impact
on autophagic flow under our experimental conditions, but data in
the literature highlighted that NLRP3 activation might be essential
to maintain pericytes in the healthy brain. Further experiments will
be needed to understand the relationship between autophagy and
inflammation in pericytes.
Acknowledgement
This work has benefited from the facilities and expertise of
PREBIOS platform (University of Poitiers, France). The authors
thank Damien Chassaing (EA3808 Neurovascular Unit and Cognitive
Disorders, University of Poitiers) for his technical skills and the
Fondation Claude Pompidou for financialFrançois A, Terro F, Janet
T, Bilan AR, Paccalin M, et al. (2013) Involvement of interleukin-
1β in the autophagic process of microglia: relevance to Alzheimer’s
disease. J Neuroinflammation 10: 915.
References
Citation
Julie V, Vincent T, Hanitriniaina R, Benjamin F, Thierry F, et al. Autophagy Monitoring in Cerebral Pericytes from Alzheimer’s disease Mouse
Model in an Inflammatory Environment. J Parkinson’s Dis Alzheimer’s Dis. 2022;9(1): 7.