
Journal of Proteomics & Computational Biology
Download PDF
Research Article
*Address for Correspondence: Mamun Mia, Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh, Tel: +8801749502149 E-mail: mamunmiabge2010@gmail.com
Citation: Mahmud S, Ahmed S, Mia M, Islam S, Rahman T. Homology Modelling, Bioinformatics Analysis and Insilico Functional Annotation of an AntitoxinProtein from Streptomyces coelicolor A3 (2). J Proteomics Computational Biol. 2016;2(1): 7
Copyright © 2016 Mahmud S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Proteomics & Computational Biology | Volume: 2, Issue: 1
Submission: 11 March, 2016 | Accepted: 19 March, 2016 | Published: 24 March, 2016
2016Reviewed & Approved by: Dr. Bin Deng, Department of Biology, University of Vermont, USA
 Subcellular localization is an indispensable feature of a protein. Cellular functions are usually localized in specific enclosed area; so, foretelling the subcellular localization of an unknown protein may possibly use to obtain handy information about their function. Therefore, this information is also valuable for the drug designing and further study about the protein [41]. Here, the subcellular localization of our targeted protein (SCO2235) predicted by CELLO is cytoplasmThe BLASTP search against non-redundant database showed a higher homology with antitoxin proteins from different Streptomyces species and showed highest of 98% homology with the target protein (Table 2). Phylogenetic analysis was depicted in the Figure 1, by using the same data. The output of the tree with the true distance guided us about the evolutionary similarity of different antitoxin genes as well as proteins.
Subcellular localization is an indispensable feature of a protein. Cellular functions are usually localized in specific enclosed area; so, foretelling the subcellular localization of an unknown protein may possibly use to obtain handy information about their function. Therefore, this information is also valuable for the drug designing and further study about the protein [41]. Here, the subcellular localization of our targeted protein (SCO2235) predicted by CELLO is cytoplasmThe BLASTP search against non-redundant database showed a higher homology with antitoxin proteins from different Streptomyces species and showed highest of 98% homology with the target protein (Table 2). Phylogenetic analysis was depicted in the Figure 1, by using the same data. The output of the tree with the true distance guided us about the evolutionary similarity of different antitoxin genes as well as proteins.
 Numerous web tools were used to search the conserved domains and potential function of SCO2235. Based on consensus predictions made by Pfam, NCBI-CDD and SUPERFAMILY suggested that the protein SCO2235 contains PhdYeFM antitox superfamily domains and is currently classified as antitoxin PhdYefM, type II toxinantitoxin system. Pfam server predicted the Antitoxin PhdYefM, type II toxin-antitoxin system at 2-72 amino acid residues with and e-value of 1.6e-18. The PhdYeFM antitox super family was also found in NCBI-CDD server at 2-81 amino acid residues with an e-value of 2.71e-22. In the SUPERFAMILY server, the domain was found at 3-79 amino acid residues with an e-value of 2.88e-22. In this system once antitoxin protein bound to their toxin companions, they can bind DNA via the N-terminus and inhibit the expression of the operons which contain genes encoding TA system [42,43].MSA of different antitoxin protein of Streptomyces and our targeted protein (gi|21220706) are depicted in the Figure 2. The secondary structure of the proteins are also included in this figure and showed that they are mostly conserved throughout the alignment along with the template.
Numerous web tools were used to search the conserved domains and potential function of SCO2235. Based on consensus predictions made by Pfam, NCBI-CDD and SUPERFAMILY suggested that the protein SCO2235 contains PhdYeFM antitox superfamily domains and is currently classified as antitoxin PhdYefM, type II toxinantitoxin system. Pfam server predicted the Antitoxin PhdYefM, type II toxin-antitoxin system at 2-72 amino acid residues with and e-value of 1.6e-18. The PhdYeFM antitox super family was also found in NCBI-CDD server at 2-81 amino acid residues with an e-value of 2.71e-22. In the SUPERFAMILY server, the domain was found at 3-79 amino acid residues with an e-value of 2.88e-22. In this system once antitoxin protein bound to their toxin companions, they can bind DNA via the N-terminus and inhibit the expression of the operons which contain genes encoding TA system [42,43].MSA of different antitoxin protein of Streptomyces and our targeted protein (gi|21220706) are depicted in the Figure 2. The secondary structure of the proteins are also included in this figure and showed that they are mostly conserved throughout the alignment along with the template.
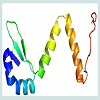
 Homology modelling is an indispensable part of the structural genomics in the recent past for the comparative modelling of various unknown structure with enormous tools [44,45]. In this study our targeted protein SCO2235, does not possess any solved crystal structure and, we predicted the comparative three-dimensional model of our protein through homology modelling. That is depicted in the Figure 3. Here, the template 3D55: A, is M. tuberculosis YefM antitoxin and showed higher amount of similarity with our target.
Homology modelling is an indispensable part of the structural genomics in the recent past for the comparative modelling of various unknown structure with enormous tools [44,45]. In this study our targeted protein SCO2235, does not possess any solved crystal structure and, we predicted the comparative three-dimensional model of our protein through homology modelling. That is depicted in the Figure 3. Here, the template 3D55: A, is M. tuberculosis YefM antitoxin and showed higher amount of similarity with our target.
 Quality assessment of the predicted three-dimensional model was acquired from PROCHECK through “Ramachandran plot” where we got 96.2% amino acid residues were within the most favored region (Figure 4 and and Table S1). The quality of our model was further checked by QMEAN6 server where the model was placed inside the dark grey zone and considered as a good model with a QMEAN6 score of 0.608 (Figure S1).
Quality assessment of the predicted three-dimensional model was acquired from PROCHECK through “Ramachandran plot” where we got 96.2% amino acid residues were within the most favored region (Figure 4 and and Table S1). The quality of our model was further checked by QMEAN6 server where the model was placed inside the dark grey zone and considered as a good model with a QMEAN6 score of 0.608 (Figure S1).

 Superimposition between the model and the template is shown in the Figure 5A. The RMSD value indicates the degree to which both the template and query structures are similar. The lower value indicates more structural similarity.
Superimposition between the model and the template is shown in the Figure 5A. The RMSD value indicates the degree to which both the template and query structures are similar. The lower value indicates more structural similarity.
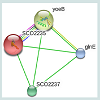
 The RMSD value obtained from the superimposition of SCO2235 and the template (3D55: A) in UCSF Chimera was found to be 0.343 Å, suggesting a reliable three-dimensional model. The Z score.Protein-protein interaction analysis from STRING database for our targeted protein is shown in the Figure 6. From the analysis, it is quite clear about the function of our targeted protein and it might be an antitoxin. The functional partners were yoeB, SCO2237 and glnE. All of these partners are associated with the TA systems.
The RMSD value obtained from the superimposition of SCO2235 and the template (3D55: A) in UCSF Chimera was found to be 0.343 Å, suggesting a reliable three-dimensional model. The Z score.Protein-protein interaction analysis from STRING database for our targeted protein is shown in the Figure 6. From the analysis, it is quite clear about the function of our targeted protein and it might be an antitoxin. The functional partners were yoeB, SCO2237 and glnE. All of these partners are associated with the TA systems.
 The active site of the protein was analyzed by CASTp server. The identification and characterization of functional sites on proteins have increasingly become an area of interest. On account of the analysis of the active site residues for the binding of ligands provides insight towards the design of inhibitors of an enzyme. In this study, we have also analyzed the best active site area of the experimental protein as well as the number of amino acid involved in it (Figure 7). Most of the cases, for the class II antitoxin have two domains, one is DNA-binding domain located in the N- terminal region and other is toxin binding domain located in the C- terminal end [46-49]. In our analysis, we have also found similar domain based active sites in our modelled protein. Those were depicted as spherical view in the Figure 7.
The active site of the protein was analyzed by CASTp server. The identification and characterization of functional sites on proteins have increasingly become an area of interest. On account of the analysis of the active site residues for the binding of ligands provides insight towards the design of inhibitors of an enzyme. In this study, we have also analyzed the best active site area of the experimental protein as well as the number of amino acid involved in it (Figure 7). Most of the cases, for the class II antitoxin have two domains, one is DNA-binding domain located in the N- terminal region and other is toxin binding domain located in the C- terminal end [46-49]. In our analysis, we have also found similar domain based active sites in our modelled protein. Those were depicted as spherical view in the Figure 7.

Homology Modelling,Bioinformatics Analysis and Insilico Functional Annotation of an Antitoxin Protein from Streptomyces coelicolor A3 (2)
Shahin Mahmud1, Md Shahabuddin Ahmed1, Mamun Mia1*, Safaiatul Islam1 and Rahman2
- 11Department of Biotechnology and Genetic Engineering, Mzzzawlana Bhashani Science and Technology University, Bangladesh
- 22Department of Biochemistry and Molecular Biology, Jahangirnagar University, Bangladesh
*Address for Correspondence: Mamun Mia, Department of Biotechnology and Genetic Engineering, Mawlana Bhashani Science and Technology University, Santosh, Tangail-1902, Bangladesh, Tel: +8801749502149 E-mail: mamunmiabge2010@gmail.com
Citation: Mahmud S, Ahmed S, Mia M, Islam S, Rahman T. Homology Modelling, Bioinformatics Analysis and Insilico Functional Annotation of an AntitoxinProtein from Streptomyces coelicolor A3 (2). J Proteomics Computational Biol. 2016;2(1): 7
Copyright © 2016 Mahmud S, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Proteomics & Computational Biology | Volume: 2, Issue: 1
Submission: 11 March, 2016 | Accepted: 19 March, 2016 | Published: 24 March, 2016
2016Reviewed & Approved by: Dr. Bin Deng, Department of Biology, University of Vermont, USA
Abstract
Streptomyces coelicolor A3 (2) is a soil-dwelling, filamentous bacteria and a reservoir of a wide range of natural antibiotics. A hypothetical protein SCO2235 of this bacterium, comprising of 87 residues was selected for in silico analysis. Numerous bioinformatics tools were used to predict the structure and function of this protein. Subcellular localization of the targeted protein was also predicted. Multiple sequence alignment (MSA) was used to locate the conserved residues and for the secondary structure analysis. Sequence homology was assessed against the protein data bank and non-redundant database by using BLASTP program of NCBI, which that revealed the targeted protein have similarity with different antitoxin protein. Homology model was obtained and PDB ID: 3D55: A served as a template having 71% homology with the protein SCO2235. The three-dimensional structure was predicted through Modeller. Validation of the three-dimensional structure was obtained through PROCHECK and QMEAN6 programs. Root Mean Squared Deviation (RMSD) calculation was used to detect super imposition of query and template structure associated with Z score. To get understandings about the physical and functional association of the targeted protein with others, STRING network analysis was implemented. Finally, the CASTp server was used to predict the active site of the protein. That is usually specific for toxin binding and DNA binding. To end, whole results hinted the biological function of the target protein to be an antitoxin.Keywords
Antitoxin; Homology modeling; Active-site; MSA; RMSDIntroduction
Streptomyces are soil-conquering gram-positive bacteria and member of the order of Actinomycetales [1]. Different strains of Streptomyces coelicolor produce numerous antibiotics, such as actinorhodin, methylenomycin, undecylprodigiosin and perimycin and it is also used for heterologous protein expression [2-4].Streptomyces coelicolor A3 (2) is so far the best genetically learned Streptomyces strain and become a standard organism for Streptomyces species [5]. The genome of S. coelicolor A3 (2) was sequenced in 2002 and, consists of 8,667,507 bp encoding 7,825 predicted genes, over 20 gene clusters for the synthesis of known or predicted natural products [6]. It has further expanded the knowledge of this organism and enabled large-scale analysis of transcriptome and proteome [7,8].
Toxin-antitoxin (TA) system was widely adopted in many genomes like bacteria and archaea and usually recognized as maintenance or stability mediator [8,9]. Although, the exact role of this system in the genome is not clear but acts as a sentinels against DNA lost and various stress management process like programmed cell death and antibiotic resistance [10]. According to the mode of action, this TA system has been classified into three broad Classes- Class I, II and Class III. Among them, Class II is predominant in many organisms [11].
The Class II TA system consists of two proteins called toxin and antitoxin. Where toxin is neutralized by antitoxin through direct protein-protein interaction and/or interacts with palindromic sequences within the promoters for suppressing transcription of the TA system [12-14].
Nowadays, sequencing technology has become more sophisticated and dealing with massive amount of data. Unfortunately, many of these genomes are still not fully annotated and comprise of various genes or proteins with anonymous function and structure. This is due to several limitations, such as the cost and time necessary for experimental methodologies. Hence, an alternative approach far from wet lab procedure called bioinformatics is now well established and, it uses algorithms and different logics derived from wet lab research to annotate the genome[15]. Recent time these sorts of approaches have got much popularity [16-19].
As the sequence is more unstable than the structure, here we tried to get some insights about the protein’s (SCO2235) function, through predicting the secondary and three-dimensional structure as well as the comparative proteomics and catalytic sites.
Materials and Methods
Sequence retrievalAt first, we inspected the NCBI (http://www.ncbi.nlm.nih.gov/) protein database for proteins containing antitoxin-like sequences [19].The hypothetical protein SCO2235 (gi|21220706|) of Streptomyces coelicolor A3 (2), consisting of 87 amino acid residues, was selected for the study. Then the sequence was stored as an FASTA format for further analysis.
Analysis of physicochemical properties
The ProtParam (http://web.expasy.org/protparam/) tool of ExPASy was used for the analysis of the physiological and chemical properties of the targeted protein sequence [20]. The properties including aliphatic index (AI), GRAVY (grand average of hydropathy), extinction coefficients, isoelectric point (pI), and molecular weight were analyzed using this tool.The hypothetical protein SCO2235 (gi|21220706|) of Streptomyces coelicolor A3 (2), consisting of 87 amino acid residues, was selected for the study. Then the sequence was stored as an FASTA format for further analysis.
Subcellular localization prediction
Determination of the subcellular localization is crucial for understanding protein function and is also vital for the genome analysis. Prediction of subcellular localization of Streptomyces coelicolor A3 (2) was carried out by CELLO v.2.5 which is a multiclass support vector machine classification system [21,22].
Comparative proteomics
The BLASTP program of NCBI database (http://www.ncbi.nlm.nih.gov/) was used for searching the similarity with our protein against the non-redundant database with default parameter [23].
Then the protein (SCO2235) was analyzed for the presence of conserved domains based on sequence similarity search with close orthologous family members. For this purpose three different bioinformatics tools and databases including Proteins Families Database (Pfam), NCBI Conserved Domains Database (NCBI-CDD) and SUPERFAMILY were used [24-26]. Pfam is a database of protein families that includes their annotations and multiple sequence alignments, generated by using hidden Markov models. NCBI-CDD is a protein annotation resource that consists of a collection of wellannotated multiple sequence alignment models for ancient domains and full-length proteins. The SUPERFAMILY annotation is based on a collection of hidden Markov models, which represent structural protein domains at the SCOP superfamily level. The annotation is produced by scanning protein sequences from over completely sequenced genomes against the hidden Markov models.
Finally, the phylogeny analysis was obtained by CLC Sequence Viewer v7.0.2 (http://www.clcbio.com/) for better understanding about their comparative evolution.
Multiple sequence alignment and secondary structure analysis
To get structural and functional insights through the sequence comparison, a combined approach was implemented. We fetched several annotated antitoxin protein sequences of Streptomyces species from the NCBI Protein database and their multiple sequence alignment (MSA) with the targeted protein were obtained through the BioEdit biological sequence alignment editor [27]. After that, these aligned sequences were used for the prediction of the secondary structure by using EsPript 3.0 [28]. We used PDB ID: 3D55 as a template source for this prediction.
Homology modelling
Homology modelling was used to determine the threedimensional structure of S. coelicolor A3 (2). A BLASTP search with default parameters was performed against the Brookhaven Protein Data Bank (PDB) to find suitable templates for homology modeling [23]. PDB ID: 3D55: A, was identified as the best template based on sequence identity (71%) between query and template protein sequence. The tertiary structure was predicted by MODELLER through HHpred tools of the Max Planck Institute for Development Biology [29-31].
Model quality assessment
The quality of the predicted structure was determined by PROCHECK and QMEAN6 programs of ExPASy server of SWISSMODEL Workspace [32-34]. Furthermore, Root Mean Squared Deviation (RMSD), superimposition of query and template structure was generated by using UCSF Chimera 1.5.3 [35]. The Z score of the template and query were also assessed by ProSA-web server [36]. Finally, the proposed model and the superimposition structure were visualized by using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, and LLC) [37].
Protein-protein interaction analysis
To perform accurate functions, protein residues interact with each other in the biological systems. We used STRING (http://stringdb.org/) database for the analysis of the protein-protein interaction (PPI) of our targeted protein. This database works through physical and functional associations derived from genomic context, highthroughput experiments, co-expression and previous knowledge to predict PPI interactions. Currently, this database covers 5, 214, 234 proteins from 1133 organisms [38].
Active site determination
Active site of the protein was determined by the computed atlas of surface topography of proteins (CASTp) server, which provides an online resource for locating, delineating, and measuring concave surface regions on three-dimensional structures of proteins [39].
Results and Discussion
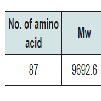
Various physiological and chemical properties of the hypothetical protein SCO2235 were assessed by ProtParam tool (Table 1). These are including aliphatic index (AI), instability index (II), pI, extinction coefficient and average hydropathicity. All of these calculations are related to the stability of the protein and that are correlates with proper function [40].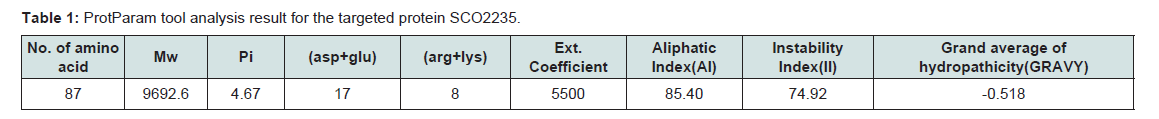
Table 1:ProtParam tool analysis result for the targeted protein SCO2235.
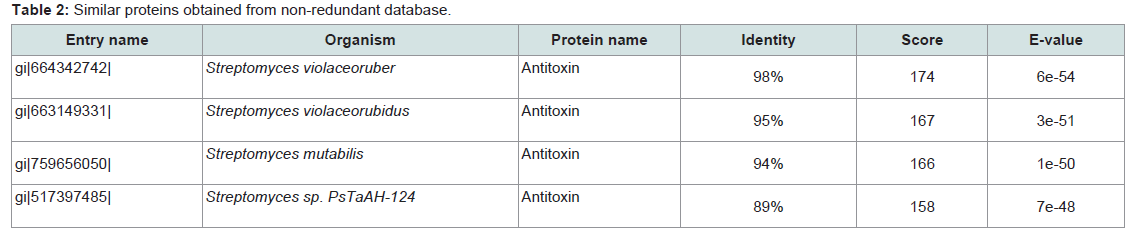
Table 2:Similar proteins obtained from non-redundant database.

Figure 1: Phylogeny analysis of different antitoxin protein of Streptomyces sp. with target protein SCO2235 (gi|212207067|) with true distance.
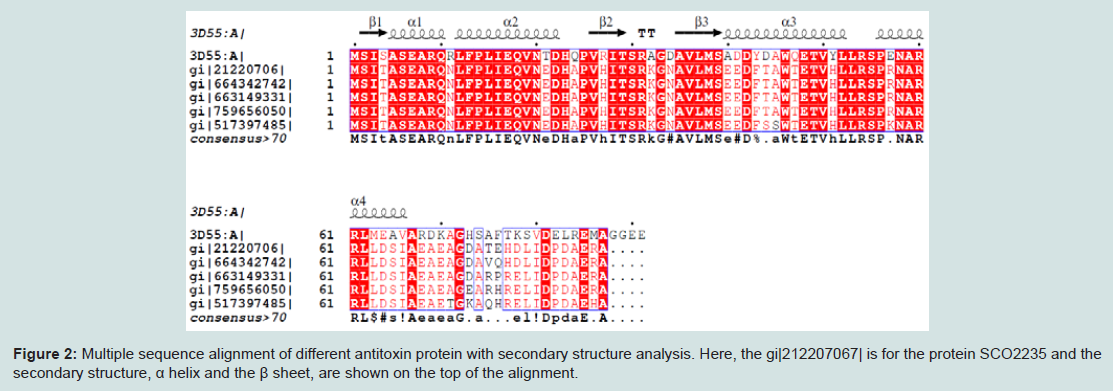
Figure 2: Multiple sequence alignment of different antitoxin protein with secondary structure analysis. Here, the gi|212207067| is for the protein SCO2235 and the secondary structure, α helix and the β sheet, are shown on the top of the alignment.
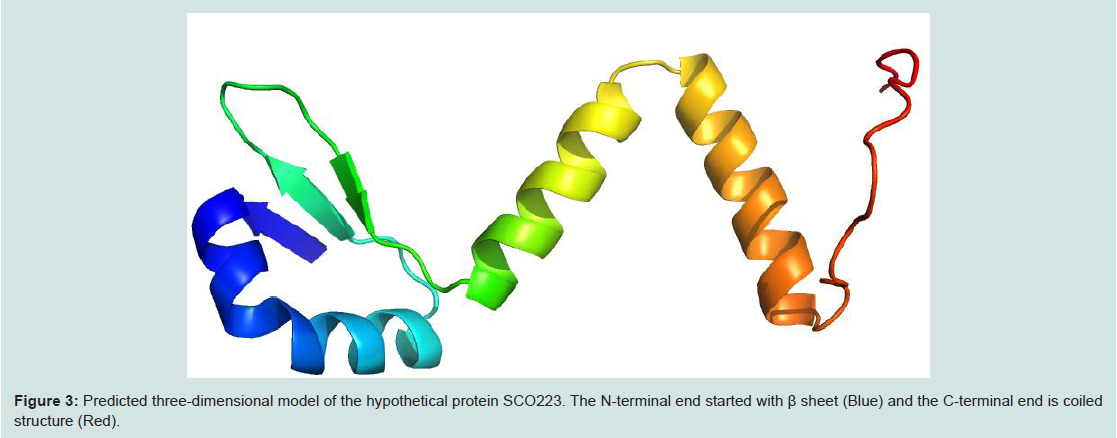
Figure 3: Predicted three-dimensional model of the hypothetical protein SCO223. The N-terminal end started with β sheet (Blue) and the C-terminal end is coiled structure (Red).
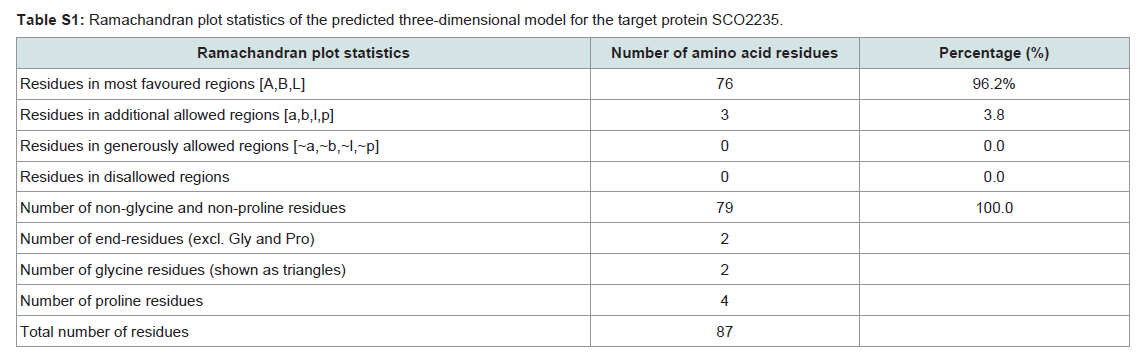
Table S1: Ramachandran plot statistics of the predicted three-dimensional model for the target protein SCO2235.
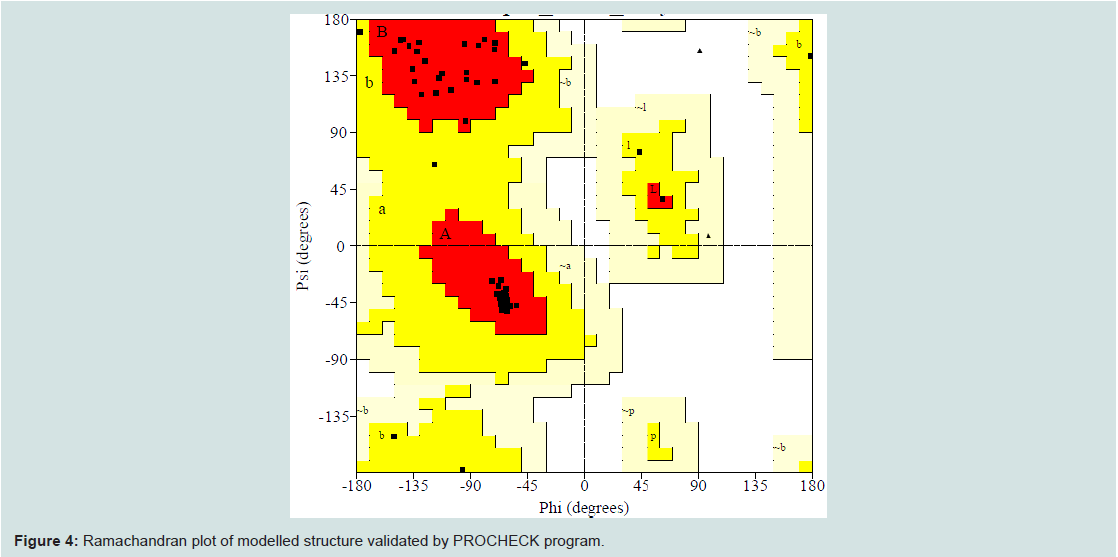
Figure 4: Ramachandran plot of modelled structure validated by PROCHECK program.
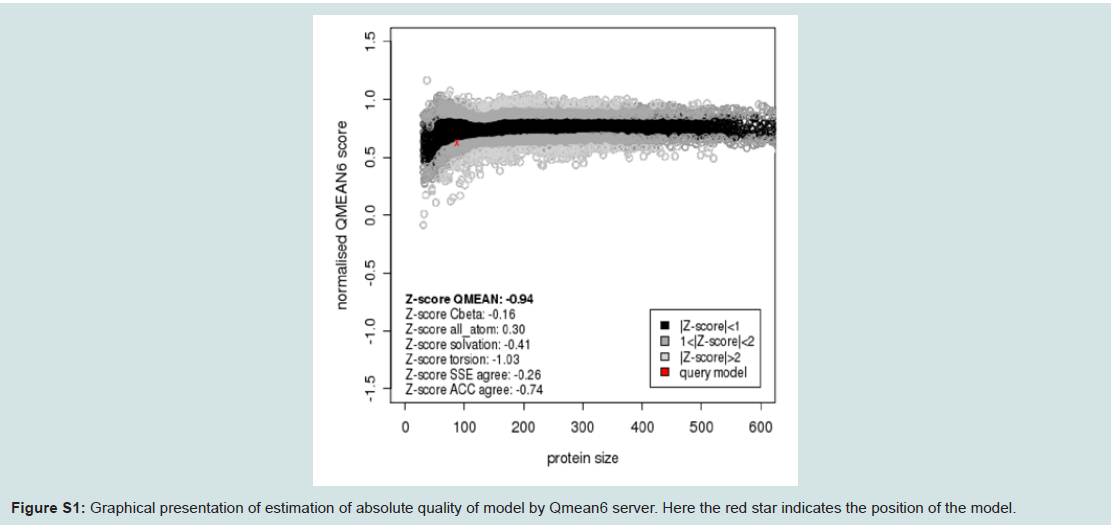
Figure S1: Graphical presentation of estimation of absolute quality of model by Qmean6 server. Here the red star indicates the position of the model.
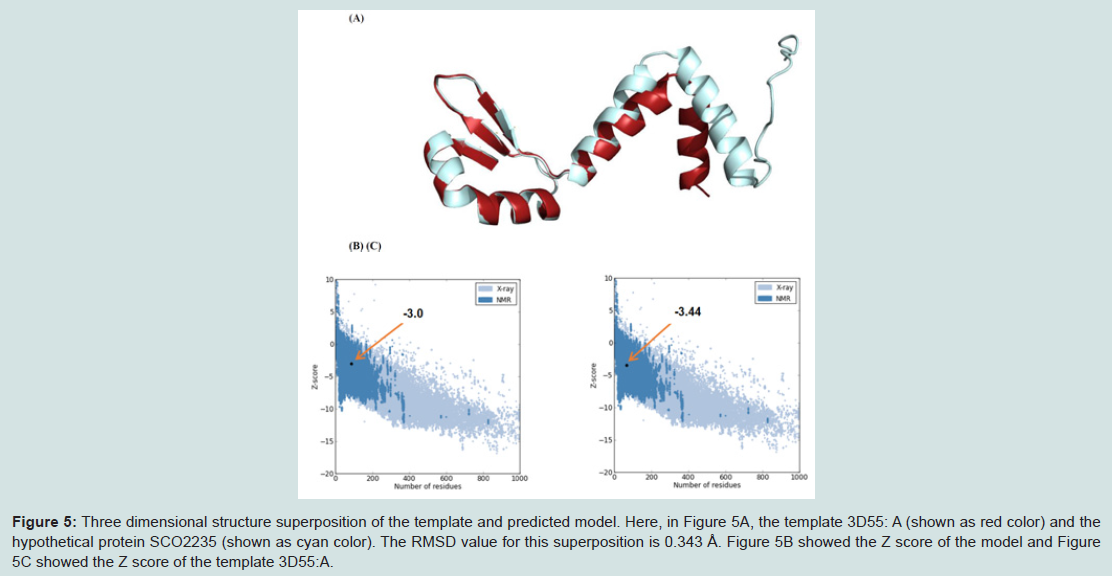
Figure 5: Three dimensional structure superposition of the template and predicted model. Here, in Figure 5A, the template 3D55: A (shown as red color) and the hypothetical protein SCO2235 (shown as cyan color). The RMSD value for this superposition is 0.343 Å. Figure 5B showed the Z score of the model and Figure 5C showed the Z score of the template 3D55:A.
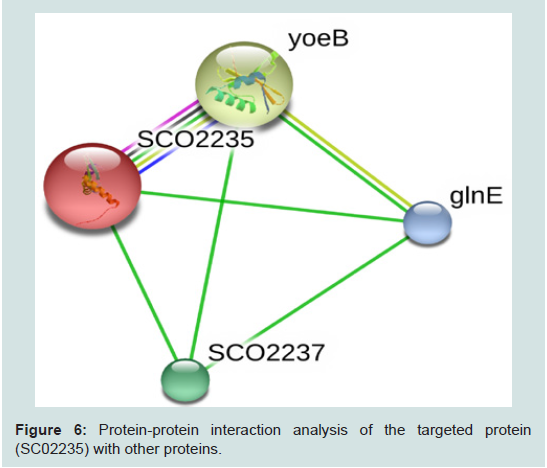
Figure 6: Protein-protein interaction analysis of the targeted protein (SC02235) with other proteins.
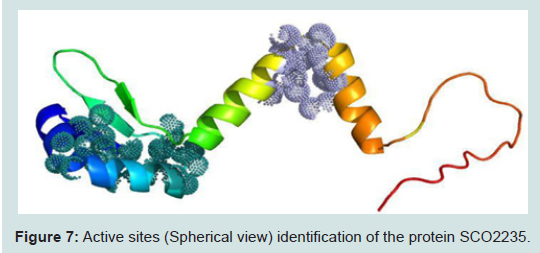
Figure 7: Active sites (Spherical view) identification of the protein SCO2235.
Conclusion
We have used homology modelling and comparative proteomics approach to predict the three-dimensional structure and possible functions for the Streptomyces coelicolor A3 (2) hypothetical protein SCO2235. With the assistance of a clearly expressed structure and annotations, we can foretell protein functional and binding sites, which can help in understanding what biological role it fulfills. All the above findings suggested that the function of the target protein is “antitoxin” which acts as type II TA system. Hopefully, this comprehensive study on this track might produce some breakthrough leads for impending research.Acknowledgements
The authors sincerely acknowledge Arafat Rahman Oany (Biotechnology and Genetic Engineering Department) for providing the necessary suggestions and facilities throughout the study.References
- Garrity G, Boone DR, Castenholz RW (2001) Bergey’s manual of systematic bacteriology. (2nd edn), Springer-Verlag New York, New York, USA.
- Brian P, Riggle PJ, Santos RA, Champness WC (1996) Global negative regulation of Streptomyces coelicolor antibiotic synthesis mediated by an absA-encoded putative signal transduction system. J Bacteriol 178: 3221-3231.
- Liu CM, McDaniel LE, Schaffner CP (1972) Fungimycin, biogenesis of its aromatic moiety. J Antibiot (Tokyo) 25: 187-188.
- http://www.sciencedirect.com/science/article/pii/S0040402001827708.
- Hopwood DA (1999) Forty years of genetics with Streptomyces: from in vivo through in vitro to in silico. Microbiology 145 (Pt 9): 2183-2202.
- Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, et al. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2). Nature 417: 141-147.
- Bucca G, Brassington AM, Hotchkiss G, Mersinias V, Smith CP (2003) Negative feedback regulation of dnaK, clpB and lon expression by the DnaK chaperone machine in Streptomyces coelicolor, identified by transcriptome and in vivo DnaK‐depletion analysis. Mol Microbiol 50: 153-166.
- Holčík M, Iyer VN (1997) Conditionally lethal genes associated with bacterial plasmids. Microbiology 143: 3403-3416.
- Van Melderen L (2010) Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol 13: 781-785.
- Lewis K (2010) Persister cells. Annu Rev Microbiol 64: 357-372.
- Makarova KS, Wolf YI, Koonin EV (2009) Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct 4: 19.
- Bailey SE, Hayes F (2009) Influence of operator site geometry on transcriptional control by the YefM-YoeB toxin-antitoxin complex. J Bacteriol 191: 762-772.
- de la Hoz AB, Ayora S, Sitkiewicz I, Fernández S, Pankiewicz R, et al. (2000) Plasmid copy-number control and better-than-random segregation genes of pSM19035 share a common regulator. Proc Natl Acad Sci U S A 97: 728-733.
- Hallez R, Geeraerts D, Sterckx Y, Mine N, Loris R, et al. (2010) New toxins homologous to ParE belonging to three‐component toxin-antitoxin systems in Escherichia coli O157:H7. Mol Microbiol 76: 719-732.
- Bhatia U, Robison K, Gilbert W (1997) Dealing with database explosion: a cautionary note. Science 276: 1724-1725.
- Oany AR, Ahmad SA, Kibria KK, Hossain MU, Jyoti TP (2014) A hypothetical protein of alteromonas macleodii AltDE1 (amad1_06475) predicted to be a cold-shock protein with RNA chaperone activity. Gene Regul Syst Bio 8: 141-147.
- Oany AR, Jyoti TP, Ahmad SA (2014) An in silico approach for characterization of an aminoglycoside antibiotic-resistant methyltransferase protein from Pyrococcus furiosus (DSM 3638). Bioinform Biol Insights 8: 65-72.
- Oany AR, Ahmed MS, Jahan N, Latif MA, Mahmud S, et al. (2015) Homology modeling and assigned functional annotation of an uncharacterized antitoxin protein from Streptomyces xinghaiensis. Bioinformation 11: 493-500.
- NCBI (2014) Hypothetical protein SCO2235 [Streptomyces coelicolor A3(2)]. NCBI reference sequence: NP_626485.1.
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, et al. (2005) Protein identification and analysis tools on the ExPASy server. In: John M, Walker, (Eds). The proteomics protocols handbook. Humana Press, ExPASy Bioinformatics Resource Portal, pp. 571-607.
- Yu CS, Lin CJ, Hwang JK (2004) Predicting subcellular localization of proteins for Gram‐negative bacteria by support vector machines based on n‐peptide compositions. Protein Sci 13: 1402-1406.
- Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64: 643-651.
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, et al. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272: 5101-5109.
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, et al. (2004) The Pfam protein families database. Nucleic Acids Res 32(Database issue): D138-D141.
- Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, et al. (2015) CDD: NCBI's conserved domain database. Nucleic Acids Res 43(Database issue): D222-D226.
- Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313: 903-919.
- Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95-98.
- Gouet P, Courcelle E (2002) ENDscript: a workflow to display sequence and structure information. Bioinformatics 18: 767-768.
- Šali A, Potterton L, Yuan F, van Vlijmen H, Karplus M (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23: 318-326.
- Söding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21: 951-960.
- Söding J, Biegert A, Lupas AN (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33(Web Server issue): W244-W248.
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26: 283-291.
- Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27: 343-350.
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22: 195-201.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, et al. (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25: 1605-1612.
- Wiederstein M, Sippl MJ (2007) ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res 35(Web Server issue): W407-W410.
- Blainebell, Herc11, Speleo (2015) The PyMOL molecular graphics system: PyMOL is an OpenGL based molecular visualization system. Sourceforge.
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, et al. (2013) STRING v9. 1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41(Database issue): D808-D815.
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, et al. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 34(Web Server issue): W116-W118.
- Shoichet BK, Baase WA, Kuroki R, Matthews BW (1995) A relationship between protein stability and protein function. Proc Natl Acad Sci U S A 92: 452-456.
- Wang J, Sung WK, Krishnan A, Li KB (2005) Protein subcellular localization prediction for Gram-negative bacteria using amino acid subalphabets and a combination of multiple support vector machines. BMC Bioinformatics 6: 174.
- Anantharaman V, Aravind L (2003) New connections in the prokaryotic toxin-antitoxin network: relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol 4: R81.
- Garcia-Pino A, Balasubramanian S, Wyns L, Gazit E, De Greve H, et al. (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142: 101-111.
- Vitkup D, Melamud E, Moult J, Sander C (2001) Completeness in structural genomics. Nat Struct Biol 8: 559-566.
- Chance MR, Bresnick AR, Burley SK, Jiang JS, Lima CD, et al. (2002) Structural genomics: a pipeline for providing structures for the biologist. Protein Science 11: 723-738.
- Santos-Sierra S, Pardo-Abarrio C, Giraldo R, Dı́az-Orejas R (2006) Genetic identification of two functional regions in the antitoxin of the parD killer system of plasmid R1. FEMS Microbiol Lett 206: 115-119.
- Smith JA, Magnuson RD (2004) Modular organization of the Phd repressor/antitoxin protein. J Bacteriol 186: 2692-2698.
- Bernard P, Couturier M (1991) The 41 carboxy-terminal residues of the miniF plasmid CcdA protein are sufficient to antagonize the killer activity of the CcdB protein. Mol Gen Genet 226: 297-304.
- Brown BL, Grigoriu S, Kim Y, Arruda JM, Davenport A, et al. (2009) Three dimensional structure of the MqsR: MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog 5: e1000706.