
Journal of Pediatrics & Child Care
Download PDF
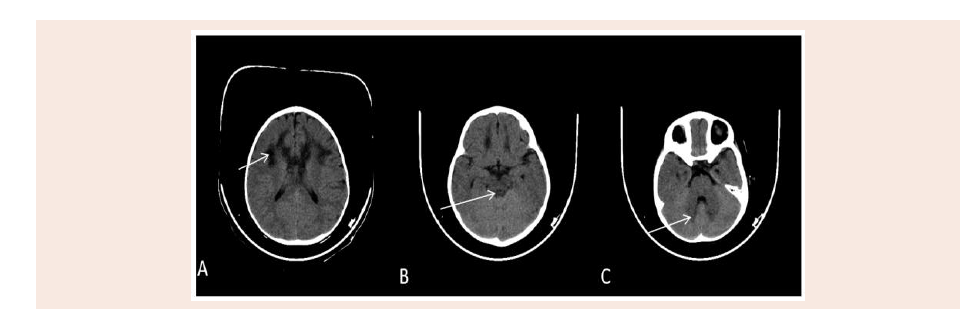 Figure 1: Axial plain CT scan of the brain show bilateral symmetrical hypo attenuating areas matching to the MRI high signal intensity at periventricular white matter
(A), periaqudect of the midbrain (B), and dentate nucleus of the cerebllum (C).
Figure 1: Axial plain CT scan of the brain show bilateral symmetrical hypo attenuating areas matching to the MRI high signal intensity at periventricular white matter
(A), periaqudect of the midbrain (B), and dentate nucleus of the cerebllum (C).
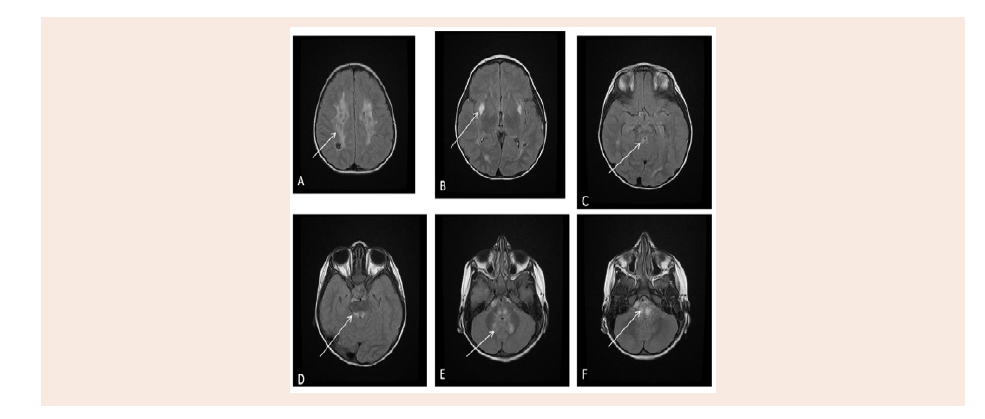 Figure 2: Axial MRI brain of different levels, T2 FLAIR pulse sequences that show bilateral symmetrical high signal intensity at periventricular white matter (A),
putamen (B), periaqueduct of midbrain (C), posterior surface of the pons (D), dentate nucleus of the cerebellum (E), and medulla oblongata (F).
Figure 2: Axial MRI brain of different levels, T2 FLAIR pulse sequences that show bilateral symmetrical high signal intensity at periventricular white matter (A),
putamen (B), periaqueduct of midbrain (C), posterior surface of the pons (D), dentate nucleus of the cerebellum (E), and medulla oblongata (F).
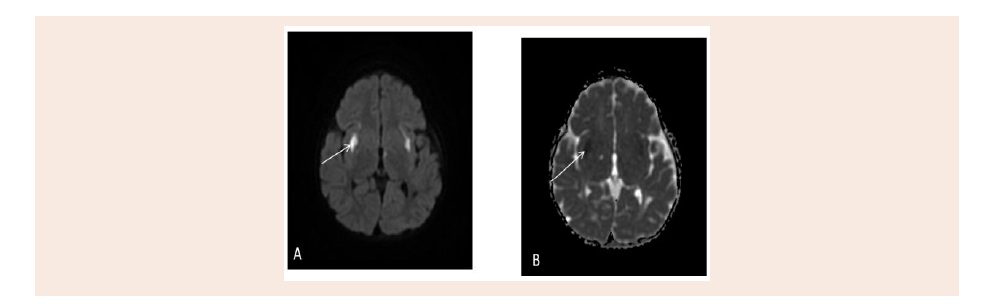 Figure 3: Active disease appear restriction as bilateral symmetrical putamen high signal intensity on Diffusion weighted image (A) and low signal intensity on
Apparent diffusion co-efficient (B).
Figure 3: Active disease appear restriction as bilateral symmetrical putamen high signal intensity on Diffusion weighted image (A) and low signal intensity on
Apparent diffusion co-efficient (B).
Case Report
Progressive Encephalopathy and Central Hypoventilation Related to Homozygosity of NDUFV1 Nuclear Gene, a Rare Mitochondrial Disease
AL-Buali MJ*, Al Ramadhan S, Al Buali H, Al-Faraj J and Al Mohanna M
Pediatric Department , Maternity Children Hospital , Saudi Arabia
*Address for Correspondence: Al-buali MJ, Pediatric Consultant and Consultant of Medical Genetics, Deputy Chairman of Medical Genetic Unite, Pediatrics Department , Maternity Children Hospital, Al-hassa, Hofuf city, Saudi Arabia; E-mail: doctormajed1@gmail.com
Submission: 15 July 2019;
Accepted: 5 August 2019;
Published: 9 August 2019
Copyright: © 2019 AL-Buali MJ, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Background:
Mitochondrial diseases are a group of disorders
caused by dysfunctional organelles that generate energy for our
body. Mitochondria small double-membrane organelles found in
every cell of the human body except red blood cells. Mitochondrial
diseases are sometimes caused by mutations in the mitochondrial DNA
that affect mitochondrial function. Other mitochondrial diseases are
caused by mutations in genes of the nuclear DNA, either as Autosomal
recessive or Autosomal dominant inheritance pattern whose gene
products are imported into the mitochondria (mitochondrial proteins)
as well as acquired mitochondrial conditions. We describe a clinical
presentation of a patient with an autosomal recessive mitochondrial
disease due to a homozygous mutation in the nuclear gene, NDUFV1
gene (OMIM: 618225).
Case presentation:
In the present study, we report 36 months
old girl from Saudi origin product of consanguineous marriage. With
the clinical presentation of failure to gain normal developmental
milestones, neuromotor regression, frequent attacks of the unexplained
decreased level of consciousness and encephalopathy associated
with central hypoventilation. There is a strong family history of similar
presentation with early childhood deaths in two other siblings with no
healthy kids for the couple. The girl evaluated thoroughly to reach a
specific diagnosis, including clinical, radiological and biochemical
work-up. However, we got an explanation for this phenotype through
molecular genetic testing and couple referred for the Preimplantation
Genetic Diagnosis.
Result:
The constellation of clinical presentation and radiological
finding confirmed by a Molecular test showed a homozygous missense
mutation c. 1268C>T p. (Thr423Met) in the NDUFV1 gene (OMIM:
618225) which is consistent with autosomal recessive mitochondrial
disease.
Keywords
Progressive encephalopathy; Central hypoventilation;
Nuclear mitochondrial disease; NDUFV1 gene
Introduction
Mitochondrial diseases are a group of disorders caused by
dysfunctional mitochondria, the organelles that generate energy
for our body cells. Mitochondria are small double membrane
organelles found in every cell of the human body except red blood
cells. Mitochondrial diseases are can be caused by mutations
in the mitochondrial DNA that affect mitochondrial function.
Other mitochondrial diseases are caused by mutations in genes
of the nuclear DNA, either as Autosomal recessive or Autosomal
dominant inheritance pattern whose gene products are imported
into the mitochondria (mitochondrial proteins) as well as acquired
mitochondrial conditions due to adverse effects of drugs, infections,
or other environmental causes [1]. Mitochondrial disease is one of the most common groups of genetic diseases with a minimum
prevalence of greater than 1 in 5000 in adults. Mitochondrial diseases
can be present at birth but can be manifested also at any age [2].
Whilst multi-system involvement is often evident, a neurological
manifestation is the principal presentation in most cases. The
multiple clinical phenotypes and the involvement of both the
mitochondrial and nuclear genome make mitochondrial disease,
particularly challenging for the clinician [3]. The clinical features
are heterogeneous and often can mimic many neurological or other
systemic diseases. The pediatric onset disease is associated with more
severe multi-systemic involvement, relentless progression and poorer
prognosis, however, there are rare exceptions, such as reversible
respiratory chain deficiency caused by them. 14674T>C mutation [4].
Most mitochondrial function and biogenesis are controlled by
nuclear DNA. Human mitochondrial DNA encodes 13 proteins of
the respiratory chain, while most of the estimated 1,500 proteins
and components targeted to mitochondria are nuclear-encoded.
Defects in nuclear-encoded mitochondrial genes are associated
with hundreds of clinical disease phenotypes including anemia,
dementia, hypertension, lymphoma, retinopathy, seizures, and
neurodevelopmental disorders [5].
NADH dehydrogenase [ubiquinone] flavoprotein 1,
mitochondrial (NDUFV1) is an enzyme that in humans is encoded by
the NDUFV1 gene. The NDUFV1 gene encodes the 51-kD subunit of
complex I (NADH: ubiquinone oxidoreductase) of the mitochondrial
respiratory chain. Defects in complex I are a common cause of
mitochondrial dysfunction. Mitochondrial complex I deficiency is
linked to myopathies, encephalomyopathies and neurodegenerative
disorders such as Parkinson’s disease and Leigh syndrome [6].
NDUFV1 is located on the q arm of chromosome 11 in position
13.2 and has 10 exons. The NDUFV1 gene produces a 50.8 kDa
protein composed of 464 amino acids [6,7].
Complex I am composed of 45 different subunits. NDUFV1 is a
component of the Flavoprotein-Sulfur (FP) fragment of the enzyme.
NDUFV1 is an oxidoreductase and a core subunit of complex I that
is thought to be required for assembly and catalysis. It is a peripheral membrane protein located on the matrix side of the mitochondrion
inner membrane [8]. Mutations in the NDUFV1 gene are associated
with Mitochondrial Complex I deficiency, which is autosomal
recessive. This deficiency is the most common enzymatic defect of the
oxidative phosphorylation disorders [9,10]. Mitochondrial complex I
deficiency shows extreme genetic heterogeneity and can be caused by
a mutation in nuclear-encoded genes or in mitochondrial-encoded
genes. There are no obvious genotype-phenotype correlations and
inference of the underlying basis from the clinical or biochemical
presentation is difficult, if not impossible. However, the majority of
cases are caused by mutations in nuclear-encoded genes. It causes
a wide range of clinical disorders, ranging from the lethal neonatal
disease in adult-onset neurodegenerative disorders. Phenotypes
include macrocephaly with progressive Leukodystrophy, nonspecific
encephalopathy, hypertrophic cardiomyopathy, myopathy, liver
disease, Leigh syndrome, Leber hereditary optic neuropathy, and
some forms of Parkinson disease. Clinical manifestations can include
lactic acidosis, cerebral degeneration, ophthalmoplegia, ataxia,
spasticity, and distortion resulting from mutations in NDUFV1 [11-14].
Here, we describe a clinical presentation of a patient with a rare
autosomal recessive mitochondrial disease due to a homozygous
mutation in the NDUFV1 gene, one of the nuclear encoded genes
that code for mitochondrial components.
Method
Human subject:
In the present study, A retrospective chart review was conducted
as well, we clinically investigated affected individually (proband)
from Saudi origin family. The proband underwent a comprehensive
clinical evaluation by a general pediatrician, radiologist, neurologist
and clinical geneticist.
Molecular genetic test:
Analysis: More than 20.000 genes in the human genome were
enriched using Roche/NlmbleGen technology (SeqCap MedExome
Library) and sequenced on an alumina HISeq 1600 system (whole
exome sequencing, WES) (details of the method at the end of the
report). The aberration In the NDUFV1 gene was identified by filtering
the exam data for homozygous variants, bioinformatically extracted
HBO (homoz. ygosly-by-descent) regions and by a literature-based
survey against the Indication of Interest.
Disturbances of mitochondrial energy metabolism occurs
with an estimated incidence of 1 In 10,000 live births and are often
caused by isolating mitochondrial complex I (NADH: ubiquinone
oxidoreductase) deficiency, which causes a wide range of clinical
disorders, ranging from the lethal neonatal disease in adult-onset
Neuro degenerative disorders. Mitochondrial complex I deficiency
shows extreme genetic heterogeneity. However, the genetic defects
are thought to be mainly of nuclear origin, especially if the symptoms
begin during Infancy. Mutations in the NDUFV1 gene are associated
with Leukodystrophy and myoclonic epilepsy, which Is inherited In
an autosomal-recessive manner.
Interpretation:
WES revealed a homozygous C>T transition at
the position. 1268 in exon 9 of the NDUFV1 gene (c. 1268C>T). This missense mutation, which was confirmed by conventional Sanger
sequencing, results in an amino acid exchange from threonine to
methionine at position p. 423 (p. Thr423Met). An allele frequency in
the general population has not been documented (ExAC), Allusedbio
informatic programs (10/10) predict this alteration to be pathogenic.
The mutation has already been described in two siblings, who carry
the mutation in a compound heterozygous state with a nonsense
mutation in exon 3 of the other allele (c. 175C>T; p. Arg59•). Both
children presented at the age of five months with repeated vomiting
and developed strabismus, progressive muscular hypotonia,
myoclonic epilepsy and psychomotor regression. A cranial CT-Scan
revealed brain atrophy. The boys died at 14 and 17 months from
aspiration pneumonia (3). In summary, the homozygous missense
mutation c.1268C>T (p.Thr423Met) In the NDUFV1 gene is probably
responsible for the clinical phenotype of Aljubarah Abdulmalek.
In the case of parental consanguinity it is very likely that the
mutation is indeed homozygous. To distinguish between homozygosity
and hemizygoslty of the mutations. 1268C>T (p.Thr423Met) with a
large deletion on the other allele were commended analysis of both
parents for the mutation. However, both scenarios are in accordance
with the clinical phenotype of your patient. Targeted molecular
genetic testing can be offered to further affected and unaffected family
members of the patient.
Method:
Genomic DNA was fragmented, and the exons of the
known genes in the human genome as well as the corresponding
exon-In iron boundaries were enriched using the Roche NimbleGen
capture technology (SeqCap MedExome Libraiy), amplified and
sequenced simultaneously by Ilumina technology (next-generation
sequencing, NGS) using an Ilumina HiSeq 1600 system. The target
regions were sequenced with an average coverage of 130-fold. For
about 98% of the regions of interest 15-fold coverage, for about 97%
in 20-fold coverage was obtained. NGS data analysis was performed
using Bioinformatics analysis tools as well as JSI Medical Systems
software (version 4.1.2). Identified variants and ideal Indels were
filtered against external and internal databases and filtered depending
on their allele frequency, focusing on rare variants with a Minor Allele
Frequency (MAF) of 1% or less. Nonsense, frameshift and canonical
space site variants were primarily considered likely pathogenic.
Assessment of pathogenicity of identifying non-synonymous
variants were performed using bioinformatic prediction programs
like mutation tester, Polyphen-2, Mutation Assessor, FATHMM etc.
Only those variants were considered likely pathogenic which were
predicted probably damaging by the majority of the used organisms.
Variants that have been annotated as common polymorphisms in
databases are in the literature were neglected.
Putatively pathogenic differences between the wild type sequence
(human reference genome, according to the UCSC genome browser.
hg19, GRCh37) and the patient’s sequence mentioned and interpreted
in this report were validated using Polymerase Chain Reaction
(PCR). Amplification followed by conventional Sanger sequencing.
The resulting sequence data for the NDUFV1 gene (OMIM 161015;
locus; chromosome 11q13.2) was compared to the reference sequence
NM_007103.3.
Restricted analysis:
This initial analysis step was conducted with
a homozygosity based strategy under the assumption of an autosomal recessive inheritance mode. Exam data were filtered against
homozygous variants and HBD (homozygosity-by-descent) region
extracted by Bioinformatics tools as large stretches of homozygous
regions from informative SNPs in the data set. Furthermore, filtering
against reported more allele frequencies in public databases while and
the functional prediction score was conducted. Finally the condition
in question is not evaluated. Incidental findings are not being reported
routinely.
Limitations of WES:
Whole exome sequencing is a rapidly
evolving type of analysis. WES is being carried out using resources
corresponding to the current technical and medical standards and
scientific knowledge. Although the majority of the exam is sufficiently
covered (about 90%), Some regions remained poorly covered or
maybe missed, And mutations in this region would escape detection.
Coverage of WES data is partially for below target panel sequencing
of genes for a distinct indication, mutations of the mitochondrial-
DNA and mutations in non-coding regulatory regions and deep intronic splice mutations can be missed. Due to missing specificity
in the sequence capture approach coding regions for which highly
homologous sequences exist in the genome are partially difficult to
interpret, and Sanger sequencing of this region is not being conducted
routinely
The bioinformatic analysis still has some limitations regarding
mapping, variant calling issues, detection of insertions/deletions
or database infrastructure and is steadily improving. Currently
applied analytical strategies might be hampered by failures from the
limitations and assumptions of filtering variants as for example a
large genetic heterogeneity of certain disorders, a reduced penetrance
of certain mutations or a misinterpretation of variants may generate
misleading results. Furthermore, too many candidate variants might
remind after filtering without any functions or final proof been
available at the limo of testing. Analysis and interpretation of WES
data strongly depend on the availability of clinical data and family
history. Clinical heterogeneity or incorrect diagnosis and family history may impact analysis strategies.
The knowledge about the causes or human genetic disorders
constantly improves due to the continuous identification of novel
disease genes. Repealed analysis or current exam date after a few years
time with further developing analysis options and resources might
lead to a more comprehensive and thereby differing result.
Case Report
Our present study, 36 months old girl presented with a history
of failure of gain normal developmental milestones noticed at the
age of 8 months. She was born full term via Cesarean section with
birth weight 3 kg (3rd centile). Her perinatal or postnatal history was
unremarkable. Negative history for neonatal intensive care admission
and she was discharged with mother in good condition. The patient
was doing well, with normal development until age of 8 months;
when she stopped gaining any developmental milestones. She was
able to sit without support, but she was not able to crawl or stand.
Gradually, she became hyperactive and had poor interaction over
the next couple of months. Parents reported a history of abnormal
movement in the form staring and loss of body tone for less than one
minute, which was infrequent 2-3 times per month.
Later, the patient was admitted 2 times to Pediatric Intensive Care
Unit (PICU) with a drop in the level of consciousness associated with
central hypoventilation, suspicion of meningoencephalitis was raised
so she was covered with antibiotics but all Cerebro-Spinal Fluid (CSF)
studies and cultures were normal. Her first admission was at the age
of 18 months, she kept ventilated for 3 months after the failure of
frequent extubating trials. Then the patient was able to extubate
herself accidentally as her level of consciousness improved. So, she
was discharged with her family after the return of her baseline clinical
condition.
By the time she was home , family stated (subjectively) that she
started to show some improvement in her developmental milestones,
she started to crawl and stand with support, she was able to say
two word sentences. No history suggestive of feeding problems or
chocking the girl evaluated by an ophthalmologist, which showed
convergent squint with no retinal changes or nystagmus. The Hearing
assessment was normal.
Her second admission was at the age of 36 months old, when she deteriorated again with encephalopathy and hyperventilation. She
ventilated again, but weaning from the mechanical ventilator was
unsuccessful, so she remained on a ventilator with chronic nursery
care in the PICU.
Her parents are Second-degree cousins and this is their third
child. She has a strong family history of mitochondrial disease. Her
older sister died at the age of 14 months with a similar presentation
of progressive encephalopathy before diagnosis was made. Her MRI
finding shows white matter changes. (Any Central Hypoventilation?)
The second sibling was having similar clinical presentation with white
matter changes in the brain Magnetic Resonance Image (MRI), died
at age 2 years (died of what? Any central hypoventilation?), WES
was done for him and confirming mitochondrial disease due to a
homozygous mutation of the nuclear gene NDUFV1 Parents (which
variant-? Class?? If the patient underwent genetic testing and results
showed both are heterozygous for the same mutation.
On examination at age 36 months, she was 16 kg (90th centile),
height 89 cm (25th centile) and head circumference 46 cm at
(> 5th centile). She is Normocephalic with no dysmorphism or
neurocutaneous stigmata. She had a bilateral squint. She had
generalized hypotonia, exaggerated deep tendon reflexes with positive
ankle clonus bilaterally. There was no abnormal movement. She was
connected to mechanical ventilators with low settings, fair air entry
bilaterally and clear chest. Her liver was palpable 3 cm below costal
margin liver span was around 7 cm. Cardiovascular examination was
unremarkable.
Basics work-up including complete blood count, renal and
liver function tests were normal. Her metabolic workup including
ammonia, lactate, Blood sugar and Tandem Mass Spectrometry
(TMS) were normal. CSF analysis was normal. EEG was done at age 18
months and shows generalized slowing and no positive epileptiform
activity. Brain CT showed symmetrical hypodensity involving the
white matter of the frontal lobes, periventricular area and the genu
of corpus callosum giving butterfly appearance and also noted focal
hypodensity in the white matter of the left cerebellar hemisphere
(Figure 1).
Magnetic Resonance Imaging of the brain (MRI) done at age
15 months with IV Gadolinium, shows bifrontal, Parietal, medial
temporal, periventricular, genu of the corpus callosum, medial thalami, midbrain, dorsal pons, dentate Nuclei and Periaueductal,
superior cerebellar peduncle abnormal high signal intensity with
restricted diffusion, and some them shows cystic changes. No
abnormal enhancement identified. No hydrocephalus was found
(Figure 2 and 3).
Finally, to reach a definitive diagnosis and to rule out other
possibilities of presenting phenotype and neuroradiological changes,
molecular genetics study has been done through (WES) which
showed Missense mutation c. 1268 C>T (P. The 423 Met) in axon 9
of the NDUFV1 gene in the homozygous state, which is classified as
causative for the patient phenotypes?
Discussion
The current patient is the first case to be reported from Saudi
Arabia. The broadband has the clinical presentation suggesting
of mitochondrial infancy Neuro-regression, unexplained
encephalopathy, as well as seizure disorder. Neurological
manifestations are the main phenotype features; Central
hypoventilation is the main presenting morbidity. Central
hypoventilation not explained by any other systemic disorder, it was
frequent and progressive with time necessitated keeping the patient
on mechanical ventilation.
These presentations can be manifested by many neurological
diseases in pediatrics caused by genetic cause or as a squeal
of acute acquired brain insult as perinatal asphyxia as well as
meningoencephalitis. However, the consanguinity of the parent and
family history of similar presentation with early childhood deaths are
highly suggestive of inherited genetic disease.
The radiological finding in the present study showed Brain CT
showed symmetrical hypodensity involving white matter, these
mentioned findings consisted of metachromatic Leukodystrophy,
biotin responsive basal ganglia encephalopathy, Leigh disease
differential diagnosis.
At this point of work-up, the advanced molecular cytogenetic test
is indicted. MtDNA the MtDNA gene test panel rated no significant
variants. Whole Exome Sequence has been done in the present study,
which showed the homozygous mutation in the nuclear encoded gene
NDUFV1 c. 1268C>T p. (Thr423Met) in the NDUFV1 gene (OMIM:
618225) in the proband and showed same mutation in the parent
in heterozygous pattern. From our mini review, a few cases about
NDUFV1 gene related encephalopathy have been reported in the
literature. Compound heterozygous described mutations were more
described [13,14]. Finally, WES it very useful to reach a diagnosis as
well to find an explanation to the family for the presence of more
than one sibling with this neurodevelopmental disease was given the
couple a chance for Preimplantation Genetic Diagnosis (PGD).
Conclusion
We suggest that the clinicians should consider the possibility of
mitochondrial diseases in the patient presented with unexplained
progressive encephalopathy and hyperventilation. Molecular genetics
test specifically Whole Exam Sequence (WES) is very helpful to
reach a definitive diagnosis and to rule other differential diagnosis.
Furthermore to delineate the long term medical care, the outcome
and genetic counseling.
Finally, we emphasize the need for longitudinal data, as
such information will provide a profile encompassing care
recommendation, Future research is needed in order to elucidate the
long-term outcome of these patients.
Acknowledgement
The authors would like to acknowledge the treating team as
well as the parent of the patient for their kind cooperation.
References
Citation
AL-Buali MJ, Al Ramadhan S, Al Buali H, Al-Faraj J, Al Mohanna M. Progressive Encephalopathy and Central Hypoventilation Related to
Homozygosity of NDUFV1 Nuclear Gene, a Rare Mitochondrial Disease. J Pediatr Child Care. 2019;5(1): 05.