
Download PDF
Review Article
*Address for Correspondence: Han-Rong Weng, Department of Pharmaceutical and Biomedical Sciences, The University of GeorgiaCollege of Pharmacy, 240 West Green Street, Athens, Georgia 30602, USA, Tel: 706-542-8950; E-mail: hrweng@uga.edu Citation: Dylan Warren Maixner and Han-Rong Weng (2013) The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation andPain. J Pharmaceutics Pharmacol 1: 001. Copyright © 2013 Maixner DW, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Journal of Pharmaceutics & Pharmacology | ISSN: 2327-204X | Volume: 1, Issue: 1

 Multiple groups have worked at idenfifying and characterizing the crystal structure of GSK3β. The first crystallized structure of GSK3β occurred in 2001 and provided insight into the mechanism by which the kinase is phosphorylated. The initial crystallization occurred in the unphosphorylated (apo) form of GSK3β (PDB ID: 1I09) [38]. GSK3β has a two domain kinase fold where the N-terminal end has a β-strand domain composed of 7 anti-parallel beta strands and a C-terminal end with a conserved alpha helical domain (Figure 2). A few characteristics of the kinase that have been determined from the crystal structure are that the kinase has an ATP binding site, an activation loop, and a glycine-rich loop. However, the inhibitory phosphorylation of Serine-9 is not present due to the lack of electron density being visible on the residue. The structure of GSK3β (PBD ID: 1Q5K) was later co-crystallized with the GSK3β inhibitor AR-A014418 in 2003, which provided additional insight into the mechanism of inhibition [39]. The mechanism for inhibition is further discussed in the pharmacology of GSK3 selection.
Multiple groups have worked at idenfifying and characterizing the crystal structure of GSK3β. The first crystallized structure of GSK3β occurred in 2001 and provided insight into the mechanism by which the kinase is phosphorylated. The initial crystallization occurred in the unphosphorylated (apo) form of GSK3β (PDB ID: 1I09) [38]. GSK3β has a two domain kinase fold where the N-terminal end has a β-strand domain composed of 7 anti-parallel beta strands and a C-terminal end with a conserved alpha helical domain (Figure 2). A few characteristics of the kinase that have been determined from the crystal structure are that the kinase has an ATP binding site, an activation loop, and a glycine-rich loop. However, the inhibitory phosphorylation of Serine-9 is not present due to the lack of electron density being visible on the residue. The structure of GSK3β (PBD ID: 1Q5K) was later co-crystallized with the GSK3β inhibitor AR-A014418 in 2003, which provided additional insight into the mechanism of inhibition [39]. The mechanism for inhibition is further discussed in the pharmacology of GSK3 selection.

 We further investigated possible changes of GSK3β activities in the spinal dorsal horn in neuropathic rats induced by partial sciatic nerve ligation through immune histochemical techniques. Eight days following peripheral nerve injury, we found that there is a decrease in phosphorylated GSK3β on the lesion side compared to the uninjured side and sham operated rat (Figure 6). We also demonstrated that phosphorylated and total GSK3β is localized to both astrocytes and neuronal cells (Figure 7). These data indicate that increased GSK3β activities in the spinal dorsal horn may contribute to the genesis of neuropathic pain induced by sciatic nerve injury. Together with findings by others, these data suggest that GSK3β may be a potential target for the development of analgesics for the treatment of neuropathic pain.
We further investigated possible changes of GSK3β activities in the spinal dorsal horn in neuropathic rats induced by partial sciatic nerve ligation through immune histochemical techniques. Eight days following peripheral nerve injury, we found that there is a decrease in phosphorylated GSK3β on the lesion side compared to the uninjured side and sham operated rat (Figure 6). We also demonstrated that phosphorylated and total GSK3β is localized to both astrocytes and neuronal cells (Figure 7). These data indicate that increased GSK3β activities in the spinal dorsal horn may contribute to the genesis of neuropathic pain induced by sciatic nerve injury. Together with findings by others, these data suggest that GSK3β may be a potential target for the development of analgesics for the treatment of neuropathic pain.


The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation and Pain
Dylan Warren Maixner and Han-Rong Weng*
- Department of Pharmaceutical and Biomedical Sciences, The University of Georgia College of Pharmacy, Athens, Georgia, 30606, USA
*Address for Correspondence: Han-Rong Weng, Department of Pharmaceutical and Biomedical Sciences, The University of GeorgiaCollege of Pharmacy, 240 West Green Street, Athens, Georgia 30602, USA, Tel: 706-542-8950; E-mail: hrweng@uga.edu Citation: Dylan Warren Maixner and Han-Rong Weng (2013) The Role of Glycogen Synthase Kinase 3 Beta in Neuroinflammation andPain. J Pharmaceutics Pharmacol 1: 001. Copyright © 2013 Maixner DW, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Journal of Pharmaceutics & Pharmacology | ISSN: 2327-204X | Volume: 1, Issue: 1
Abstract
Neuroinflammation is a crucial mechanism related to many neurological diseases. Extensive studies in recent years have indicated that dysregulation of Glycogen Synthase Kinase 3 Beta (GSK3β) contributes to the development and progression of these disorders through regulating the neuroinflammation processes. Inhibitors of GSK3β have been shown to be beneficial in many neuroinflammatory disease models including Alzheimer’s disease, multiple sclerosis and AIDS dementia complex. Glial activation and elevated pro-inflammation cytokines (signs of neuroinflammation) in the spinal cord have been widely recognized as a pivotal mechanism underlying the development and maintenance of many types of pathological pain. The role of GSK3β in the pathogenesis of pain has recently emerged. In this review, we will first assess the GSK3β structure, regulation, and mechanisms by which GSK3β regulates inflammation. We will then describe neuroinflammation in general and in specific types of neurological diseases and the potential beneficial effects induced byinhibiting GSK3β. Finally, we will provide new evidence linking aberrant levels of GSK3β in the development of pathological pain.Introduction
Glycogen synthase kinase 3 (GSK3) is a serine/threonine protein kinase, which is part of the mitogen activated protein (MAP) kinase family and is pivotal in many signaling cascades [1]. GSK3 is important in metabolism and signaling in development. The role of GSK3β in mediating peripheral and central nervous system inflammation in a multitude of neurological disorders has been extensively studied [2-6]. Studies of the role of GSK3β in pathological pain have recently just begun [5,7]. In the brain, GSK3β is localized primarily to neurons [8], but has also been shown to be in glial cells [9]. Inflammation of the brain has become recognized as acommon feature shared by many neurological disorders like Alzheimer’s disease [10-12], schizophrenia [13,14], multiplesclerosis [15,16], and HIV induced dementia [17,18]. Aberrantlevels or activities of GSK3 play a critical role in the development of these diseases and pharmacological inhibitionof GSK3β ameliorates these diseases [19-23]. Inflammation is also a critical component contributing to the development and maintenance of pathological pain induced by peripheral tissue or nerve injury. Accumulation of inflammatory cells including macrophages, neutrophils at the peripheral injury site and the dorsal root ganglion, proliferation and activation of microglia and astrocytes in the spinal dorsal horn, as well as the release of pro-inflammatory cytokines and other pro-inflammatory mediators in the injury site, the dorsal root ganglion and the spinal dorsal horn have all been shown to contribute to the development and maintenance of pathological pain [24-27]. Similarly, pharmacological inhibition of GSK3β has been recently shown to attenuate pathological pain induced bynerve injury or formalin injection [5,7]. In this review, we will first briefly discuss the history,structure, regulation, and pharmacology of GSK3β. We willthen provide an overview of neurological diseases including pathological pain where neuroinflammation plays a crucial roleand how GSK3β may play a role in the progression of these diseases.Brief History, Functional Properties, and Structural Insights of GSK3
Glycogen Synthase Kinase 3 (GSK3) was first purified from rabbit skeletal muscle in 1980 and subsequently classified as a kinase based on its ability to phosphorylate and inactivate Glycogen Synthase, acting as a regulator in Glycogen synthesis [28]. However, Glycogen Synthase was thought to exist as early as the 1960s [29]. This kinase was later isolated and characterized from rat skeletal muscle [30]. Three forms of Glycogen Synthase Kinase were further identified that are referred to as Glycogen Synthase Kinase 3, Glycogen Synthase Kinase 4, and Glycogen Synthase Kinase 5, which regulates Glycogen Synthase by producing different levels of phosphorylation [31]. Glycogen Synthase Kinase 5 is referred to as Casein Kinase-2 (CK2), which is a primer of Glycogen Synthase that is phosphorylated by GSK3 [32,33]. In the early 1990s, it was shown that there are two similar forms of GSK3, GSK3-alpha (GSK-3α) and GSK3-Beta (GSK-3β) [8,34]. GSK3α and GSK3β differ in their C and N terminals, however, they share 98% sequence homology in their catalytic domains resulting in 84% overall sequence homology [8]. GSK3 is a serine/threonine kinase which is constitutively active in resting cells from a variety of tissues [35,36]. GSK3 has been implicated in many cellular processes and is thought to phosphorylate over 50 substrates [6]. In the following sections, we will mainly focus on GSK3β. Through recent advances in bioinformatic approaches, we have used the web service software from pathway Linker to produce a link between GSK3β and its signaling pathways (Figure 1) [37]. In addition to the protein signaling pathways, Table 1 represents signaling pathways where GSK3β is significantly involved [37]. As can be seen in Figure 1 and Table 1, GSK3β is involved in a diverse range of signaling pathways. Some of the classic pathways involved in inflammation and pain which are represented in Table 1 are the chemokine, B cell, opioid, leukocyte, and toll-like receptor signaling pathways.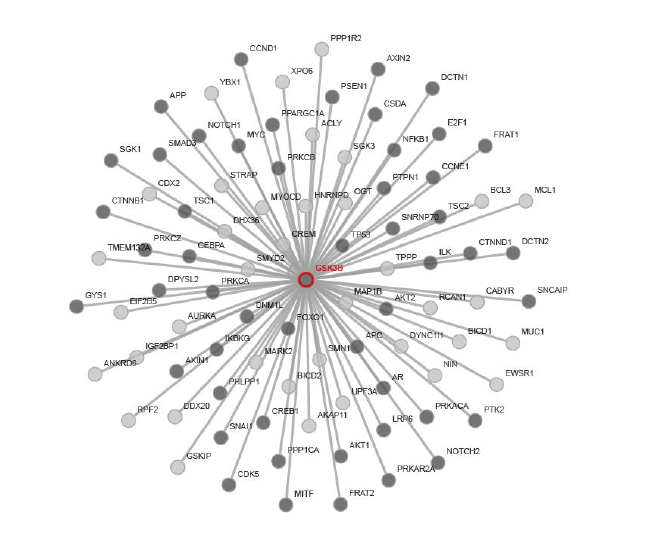
Figure 1: GSK3β and first neighbor interactions in Homo sapiens. The dark gray nodes represent interactions involved with proteins in non-signaling pathways. The lightgray nodes represent interactions with proteins in signaling pathways. Schematic and interactions of GSK3β were produced using PathwayLinker [37].
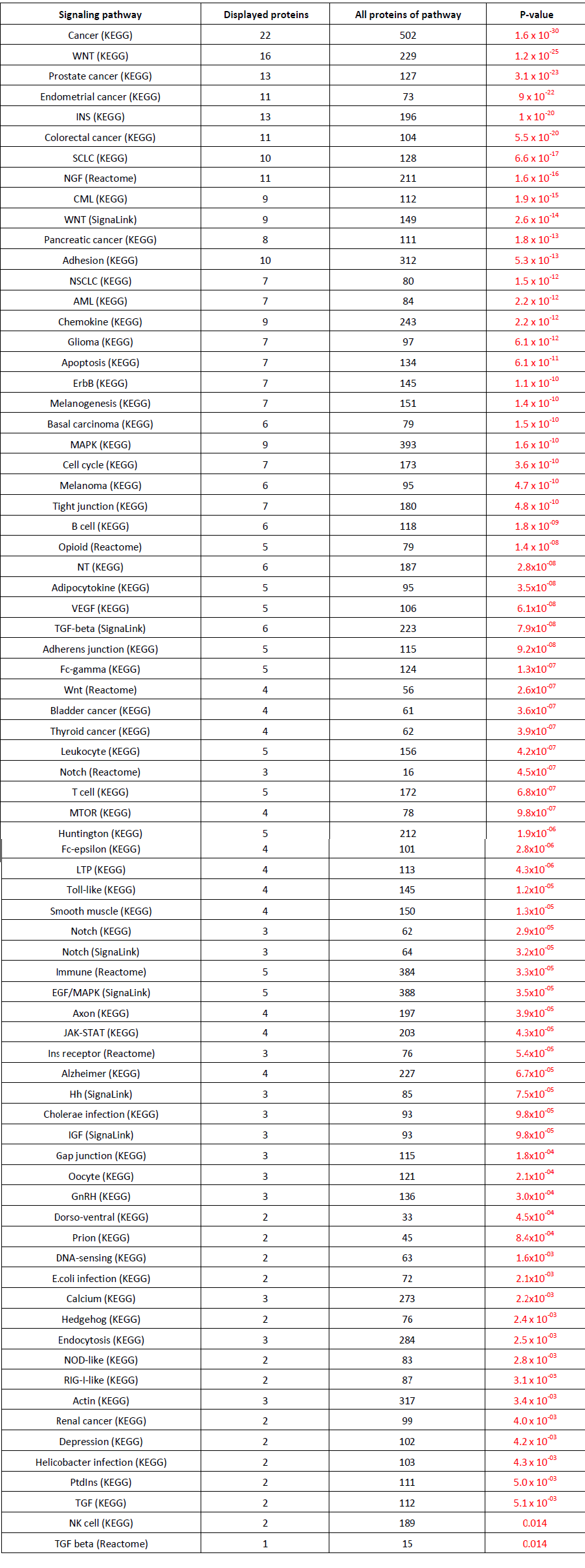
Table 1: GSK3β associated signaling pathways that are over expressed in Homo sapiens. Briefly, all first neighbor interactions of GSK3β and all proteins of Homo sapiens are queried against different signaling pathways. Overrepresentation is determined if members of the different signaling pathways are increased in the first neighbor interactions of GSK3β compared to all proteins. Signaling pathway indicates the signaling pathway along with the source. Displayed proteins indicate first neighbor interactions of GSK3β. All proteins of pathway represent the total number of proteins implicated in the signaling pathway. Pathway Linker was used to identify overexpressed signaling pathways [37].
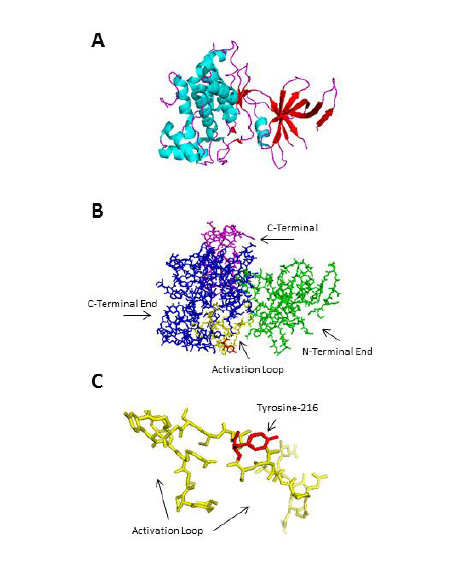
Figure 2: The crystal structure of GSK3β (Protein Data Bank PDBID: 1i09 [38]. A) The secondary structure of GSK3β showing seven anti-parallel β strands at the N-terminal end and an α-helical domain at the C-terminal end. Cyan indicates a helix, magenta indicates a loop, and red indicates a sheet. B) The overall structure of GSK3β. The N-terminal end is labeled green and encompasses residues 25-138, the C-terminal end is labeled blue and encompasses residues 139-343, and the activation loop is labeled yellow and encompasses residues 220-226 where Tyrosine-216 (red) is a phosphorylation site. C) A magnified view of Tyrosine-216, which is located in the activation loop. Figure was generated using PyMOL 1.4.1.
Regulation of GSK3
The regulation of GSK3β has been extensively studied and reviewed [40,41]. There are different ways of regulating GSK3β which occurs through phosphorylation of specific aminoacid residues, by the formation of protein complexes with GSK3β, and with pharmacological interventions. The regulation of GSK3β can occur through the phosphorylation of specific residues and the subsequent activation or inactivation of the kinase. Inactivation of GSK3β can occur through phosphorylation of Serine-9 [8,42], Serine-389 [43], Threonine-390 [43], or Threonine-43 [44], while activation can occur through the phosphorylation of Tyrosine-216 [45]. Phosphorylation of the Serine-9 residue has been extensively studied and is a major mechanism to regulate GSK3β activities by the inactivation of GSK3β. Activation of three upstream signaling pathways are known to inhibit GSK3β activity through phosphorylation of the Serine-9 residue including, the phosphatidyl inositide 3-kinases (PI3K), the mammalian target of rapamycin (mTOR), and indirectly through the mitogen-activated protein kinase/extracellular signal-regulated kinases (MAPK/ERK) pathway [41]. The signals which lead to the downstream phosphorylation and inhibition of GSK3β occurs by way of a diverse group of substances including, but not limited to, amino acids, growth factors, esters, and insulin [41]. GSK3β pathways have drawn increased attention andthe complexity of the pathways has been heavily studied in the field [3,46,47]. One of the first identified modulators of GSK3β phosphorylation was AKT, which is also termed Protein Kinase B (PKB) and is downstream of the PI3K pathway [48]. Initially,insulin was identified to inhibit GSK3β; however, the mechanism and specificity were unclear [48-50]. In studies that examined the effects of insulin on purified Glycogen Synthases, it was shown that the effect on glycogen metabolism was mediated by Glycogen Synthase 3 [35,48,51]. Glycogen synthase kinase serine residues were later identified to be dephosphorylated by insulin in vivo [51]. It was not until the identification of GSK3α and GSK3β when it was determined that insulin negatively regulates GSK3 [52]. The importance of the phosphotidylinositol (PI) 3-kinase in the signaling pathway was demonstrated by showing that inhibition of PI3K reverses insulin induced GSK3 inhibition and that incubation with serine/threonine phosphatases reverses insulin’s inhibitor effect [53]. Soon after, serine residues were identified as phosphorylation targets that mediate the inactivation of GSK3 following insulin administration which is facilitated by PKB. The serine residues which are phosphorylated and inactivated following insulin inhibition are Serine-21 for GSK3α andSerine-9 for GSK3β [48]. Two signaling pathways of GSK3β that act as regulators of the kinase are mTOR and ERK. Glycogen synthase and Glycogen synthesis were first shown to be regulated through the mTOR pathway in human muscle cells. It was also shown that amino acid availability regulates glycogen synthesis through GSK3β Serine-9 phosphorylation independently in the mTOR pathway [54]. Following the identification of mTOR as a regulator of GSK3β, S6K was shown to be downstream of mTOR and to regulate GSK3β phosphorylation [55]. These studies show that GSK3β is regulated directly by S6K differently from how it is regulated in the PI3K pathway. Following the inhibition of AKT, S6K phosphorylates the Serine-9 residue of GSK-3β in vivo [55]. Another regulator of GSK3β which entails a different signaling pathway is the ERK pathway. This pathway is different from the PI3K and mTOR pathways in that ERK regulates GSK3β through a priming mechanism. In hepatocellular carcinoma, it has been shown that GSK3β is phosphorylated at the Threonine-43 residue following ERK activation. In response to threonine-43 phosphorylation, MAPK-activated protein kinase 1 (MAPKAP-K1/p90RSK) acts to phosphorylate the primed GSK3β kinase at serine-9 residue rendering it inactive [44]. Another site of phosphorylation is the residue Tyrosine-216, which results in an increased activity of GSK3β [45] (Figure 2C). This was identified by the substitution of the tyrosine site with a phenylalanine residue showing that only Tyrosine-216 is phosphorylated under native conditions. In the catalytic domain, Tyrosine-216 lies in the activation loop and phosphorylation is needed for activity [45]. The activation loop, where catalytic activity increases following phosphorylation of Tyrosine-216, is highlighted in Figure 2B. In Dictyostelium, it has been shown that ZAK1, a tyrosine kinase, activates and phosphorylates GSK3β in vitro [56]. Phosphorylation of Tyrosine- 216 has been shown to be increased following nerve growth factor (NGF) withdrawal and protein kinase C inhibition with staurosporine in vitro [57]. In a rat ischemic model, Tyrosine-216 phosphorylation is increased [57]. Lysophosphatidic acid (LPA) has been shown to phosphorylate Tyrosine-216 during neurite retraction [58]. Tyrosine-216 phosphorylation has been shown to occur through phospholipase C activation of Protein Tyrosine Kinase 2 Beta (Pyk2), which phosphorylates the tyrosine on GSK3β and microtubule-associated proteins [58]. The upstream signaling pathways of Tyrosine-216 phosphorylation have been minimally studied compared to Serine-9 inhibition, and these studies indicate that tyrosine activation may play a role in the regulation of GSK3β; however, further studies are needed to examine the role of tyrosine phosphorylation in disease progression. GSK3β can also be regulated through the formation of protein complexes which occurs in both the Wnt and Hedgehog pathways [60,61]. The Wnt pathway has been shown to play a role in tissue maintenance, cell-cell interactions, and dysfunction in signaling may lead to degenerative diseases [46]. Proteins which bind with GSK3β to form an active or inactive complex are Axin and GSK3 binding protein (GBP), respectively [62]. In Xenopus embryos, it has been shown that the Wnt pathway is negatively regulated by the presence of Axin [63]. Through protein isolated from a rat cDNA library, it has been determined, in vitro, that the interaction between GSK3β and beta-catenin is negatively regulated through Axin binding to GSK3β in the Wnt pathway [62]. Axin is stabilized by the phosphorylation of GSK3β, which causes an increase of Axin [64]. Briefly, GSK3 forms a complex with Axin and adenomatous polyposis coli (APC), both substrates of GSK3, which phosphorylate beta-catenin leading to degradation in the absence of an initial Wnt ligand [65-67]. However, when a Wnt ligand is present, GSK3 does not form a complex with Axin and active GSK3 inhibits phosphorylation of beta-catenin preventing degradation of beta-catenin through the proteasome pathway [68,69]. These studies indicate the importance of GSK3β complex formation in the Wnt pathway and the role of GSK3β inhibition on signaling [70]. The Hedgehog pathway is important in both normal development and the production of cancers [71,72]. GSK3 has been shown to be a negative regulator of the Hedgehog pathway in Drosophila through forming a complex with Protein Kinase A (PKA) for proteolytic processing of the DNA-binding protein, Cubutis interruptus (Ci) [61]. Briefly, a complex forms between PKA, GSK3, and casein kinase 1 (CK1) which binds to costal-2 (Cos2). Following binding to Cos2, the complex phosphorylates Ci causing an inhibitory effect on the pathway due to the phosphorylation [72]. The phosphorylation of Ci causes the proteolytic degradation from an active form to an inactive form [73]. The decrease in phosphorylation of Ci leads to a decrease in proteolytic processing producing an opposite effect [61]. These studies indicate the relevance of GSK3β in complex formations and further work is needed to studythe role in disease progression.Pharmacology of GSK3
Aberrant regulation of GSK3β has been implicated and studied in several disorders such as Alzheimer’s disease, type 2 diabetes, and bipolar disorder [6,74-79]. Many pharmacological agents have been developed due to the involvement of GSK3β in the different diseases with different mechanisms of action [80]. There have been six completed clinical trials involving inhibitors of GSK3 in Alzheimer’s disease [81], hair loss [82], progressive supranuclear palsy [83,84], and bipolar disorder [85]. There are currently three clinical trials which are recruiting for bipolar disorder [86,87], spinal cord injuries and muscle atrophy [88]. In addition, there is a suspended clinical trial in patients with gliomas [89]. The broad spectrum of clinical trials indicates the importance of GSK3 in many pathological processes. GSK3β activity can be regulated by pharmacological interventions. The most extensively studied GSK3β inhibitor is lithium chloride [90]. Within this context, lithium has been extensively studied as a regulator of GSK3β signaling pathways in bipolar disorder [91]. While lithium chloride has been shown to be effective in treating bipolar disorder, high concentrations are needed for physiological and pharmacological effects [92]. Lithium was first shown to have an effect on GSK3 in the early 1990s [90]. GSK3 was identified as a target in intact cells and in vitro showing the potential role of GSK3 in development [92]. In the nervous system, axonal growth has also shown to be decreased by lithium administration [93] and in a mouse model of degeneration [20]. Compared to other inhibitors, lithium chloride is a noncompetitive ATP inhibitor which competes with Mg2+ for association with the kinase and is most effective in vivo [94]. Lithium also has a high inhibitor constant, Ki = 2 mM [94,95]. Lithium compared to other inhibitors, at high doses can inhibit both GSK3β and GSK3α [96]. Also, lithium has been shown to indirectly activate AKT in neuronal cells which can consequently phosphorylate kinases other than GSK3β and has also shown to be protective against glutamate excitotoxicity in vitro [97]. These studies point to the potential clinical andtherapeutic effects of lithium and the lack of specificity for GSK3β. Two GSK3β inhibitors of interest that utilize a different mechanism of inhibition other than lithium chloride are SB216763 and AR-A014418 (Figure 3A and Figure 3B). For both inhibitors, their mechanism of action is through binding in the ATP pocket of GSK3β [98]. SB216763 is an anilinomaleimide with a Ki ranging from 10 nM to 30 nM for GSK3α and in the presence of ATP inhibits GSK3β activity by up to 96% [99]. AR-A014418 is a thiazole with a Ki of 38 nM and inhibits GSK3β with an IC50 = 104+/-27 nM. The inhibitor binds within the ATP pocket along the hinge/linker region where the nitro group engages the ATP pocket [39]. GSK3α is also inhibited by these small molecule inhibitors; however the focus of this review is on the inhibition of GSK3β. Both of these small molecule inhibitors are more selective and potent compared to lithium chloride, which may help in identifying the specific role of GSK3β in different disease pathologies.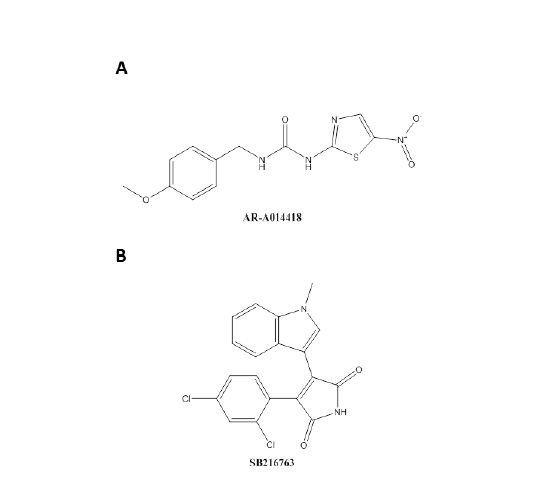
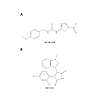
Figure 3: The chemical structure of two GSK3β inhibitors. A) The chemical structure of 1-(4-methoxybenzyl)-3-(5-nitrothiazol-2-yl) urea (AR-A014418) with the MW=308.31. B) The chemical structure of 3-(2,4-dichlorophenyl)-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione (SB216763) with the MW=371.22 [39,99].
GSK3β Inflammatory Signaling Pathways
Glycogen Synthase Kinase 3 has been identified as a target for inflammatory mediated diseases [6] and plays a key role in mediating inflammatory responses (see Table 1 for signaling pathways). The role of GSK3β in inflammation was first shown by Martins and coworkers in 2005 [100]. They determined that following the development of inflammation, the kinase acts as a modulator for the expression of key pro-inflammatory and anti-inflammatory cytokines derived from monocytes and other peripheral blood cells to dampen inflammatory responses [100]. The mechanism by which GSK3β attenuates inflammation has been hypothesized to be regulated, in part, through the nuclear translocation of the transcriptional factor CREB (cAMP Response Element-Binding Protein) [100]. GSK3 has been shown to have an inhibitory effect on CREB regulation resulting in decreased nuclear translocation of CREB [101]. The decreased translocation of CREB into the nucleus increases the expression of pro-inflammatory cytokines such as Interleukin-1-Beta (IL-1β) and Tumor Necrosis Factor -1 alpha (TNF-α). Inhibition of GSK3β increases CREB DNA binding activity, which increases the transcription and expression of anti-inflammatory cytokines (IL-10) [100,102,103]. In dendritic cells, GSK3β is involved in TNF-α and IL-6 secretion [104]. These studies provide evidence for the importance of GSK3 activity in regulating pro- and anti-inflammatory response where by an increase in GSK3β activity increases the production of pro-inflammatory cytokines while a decrease in GSK3β activity results in the production of anti-inflammatory cytokines. The mechanism by which of GSK3 regulates CREB translocation has been further elucidated in a model for intestinal inflammation. Following the development of inflammation, inhibition of GSK3 reduces the pro-inflammatory phenotype of Toll-like receptors [100]. In addition, inhibition of GSK3 modulates the transcriptional activity of both CREB and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), a master regulator of inflammation, in the intestinal immune cells [105]. The translocation of NF-κB from the cytoplasm to the nucleus results in the transcription of pro-inflammatory genes such as IL-1β, TNF-α, and IL-6 [106]. In a model of acute inflammation, inhibition of NF-κB in leukocytes decreases inflammation [107] and adenoviral infection of human macrophages, which inhibits NF-κB activation, decreases the production of TNF-α [108]. Interleukin-10, an anti-inflammatory cytokine, decreases the transcription of NF-κB resulting in a decrease in the expression of pro-inflammatory cytokines [109]. The activation of GSK3β modulates the nuclear translocation of both NF-κB and CREB by enabling CREB Binding Protein (CBP) to bind both transcriptional factors, which facilitates nuclear translocation and increases the production and transcription of pro-inflammatory cytokines. When GSK3 is inactivated, NF-κB translocation is decreased while CREB transcription increases [101]. This result in a relative increase of CREB in the nucleus compared to NF-κB and increases the binding of CBP to CREB. The increase of CBP binding to CREB produces an increase in the transcription of anti-inflammatory cytokines (e.g., IL-10) [100,103]. The importance of GSK3 in regulating anti-inflammatory cytokines through the NF-κB pathway has been shown in GSK3 mouse knockouts [110]. In addition, it has been shown that inhibition of GSK3 can decrease NF-κB activation in hepatocytes [111]. It has also been shown in hepatocytes that the inhibition of GSK3β reduces NF-κB activity, increases CREB transcription factor, and attenuates TNF-α mediated apoptosis [101]. These studies indicate the importance of CREB and NF-κB transactivation through GSK3 in regulating the release of pro-inflammatory and anti-inflammatory cytokines (Figure 4).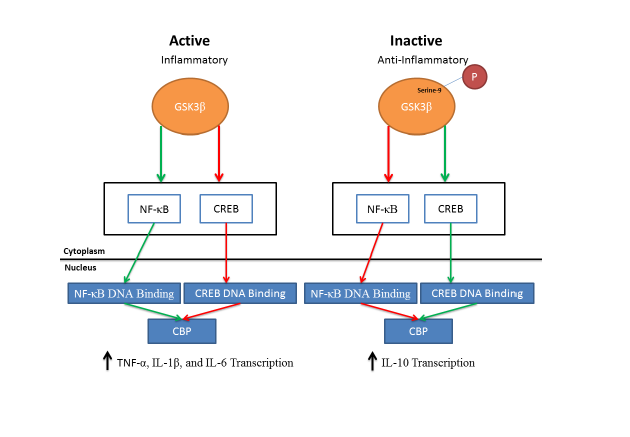
Figure 4: Putative downstream pathways of GSK3β that modulate the expression of pro-inflammatory and anti-inflammatory cytokines. Following GSK3β activation, NF-κB istranslocated from the cytoplasm to the nucleus and binds transcriptional sites with CBP leading to an increase in the transcription of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6). An increase in GSK3β phosphorylation (inhibition of GSK3β) results in an increase of CREB being translocated from the cytoplasm to the nucleus. This results in an increase in CBP binding with CREB at transcriptional sites, which increases the transcription of anti-inflammatory cytokine (IL-10). Green lines indicate an increase in activation and red lines indicate a decrease in activation.
Neuroinflammation and GSK3
Neuroinflammation is an inflammatory response that is characterized by glial activation and the production of inflammatory cytokines [112]. There are many diseases which are linked to the increased activation of glial cells and elevated pro-inflammatory cytokines. Neuroinflammation has been implicated in multiple neurological disorders of the central nervous system (CNS) such as acquired immune deficiency syndrome (AIDS), stroke, andmultiple sclerosis [113]. These disorders all share a common neuroinflammatory response; however, the etiologies of these disorders vary. GSK3β has recently been shown to be involved in the activation of glial cells. In rat cortical glia, GSK3β is expressed in both astrocytes and microglia and is activated following exposure to lipopolysaccharide (LPS) [114]. Following stimulation, pro-inflammatory cytokines are increased and inhibition of GSK3β attenuate the production of pro-inflammatory cytokines (IL-1β and TNF-α) and augments the production of anti-inflammatory cytokines (IL-10) in vitro [114]. In addition, GSK3 activation has been linked to the increase in glial cell proliferation [115]. Inflammation of the brain is isolated from the body through the Blood Brain Barrier (BBB), where an increase in immune cell trafficking occurs following injury [116,117]. Originally, the CNS was thought to be protected from systematic inflammation, however, it is now established that multiple factors can contribute to a neuroinflammatory response [118]. The inflammatory response of the brain appears to be more tightly regulated than peripheral inflammation and regulation occurs through the activation of glial cells [119]. In the CNS, glial cells encompass nearly 75% of the overall cells where the majority of glial cells are astrocytes and oligodendrocytes while the rest are microglia cells [120]. These three types of glial cells, particularly microglia and astrocytes act as immune responders in the CNS. Microglia cells were first established to be a different cell type compared to astrocytes and oligodendrocytes in the early 1900s [121]. Microglia cells act as macrophages of the nervous system in normal conditions [122]. They are considered to be resident macrophages even though they only consist of 15% of the cells in the CNS [123,124]. In the CNS, it has been shown that microglia cells become activated in different pathological conditions and release pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) and chemokines [125]. The role of microglia cells in facilitating the proliferation of immune cells has also been thoroughly studied [126,127]. Astrocytes, in regards to neuroinflammation, act as scavengers of the CNS and recycle excess neurotransmitters and ions. Dysregulation of astrocytes has been shown to play a role in the development of neurological diseases [128]. Under normal conditions, astrocytes regulate excess glutamate in the synaptic cleft preventing neurotoxicity [1269,130]. Following the accumulation of pro-inflammatory cytokines in the CNS, astrocytes become activated and proliferate. The increase in astrocytic activation causes an increase in proliferation, which can be identified with the increase in glial fibrillary acidic protein (GFAP) [131]. Although GFAP is currently used for immunostaining and Western blotting of astrocytes, it only identifies ~15% of the total volume of the astrocyte [132]. An in vitro model of metabolic injury shows that astrocytes release interleukin 1 (IL-1), IL-6, TNF-α and interferon gamma (IFN-g) [133]. When a neuroinflammatory response ensues, glial cells become activated and produce pro-inflammatory cytokines such a TNF-α, IL-1β, and IL-6 and chemokines [134,135]. Microglia cells act as the first responders to injury while astrocytes sustain the inflammatory response. Neuroinflammation can be seen as both beneficial and detrimental. Some of the common themes involved in the beneficial aspects of neuroinflammation occur through neuroprotection and axonal regeneration. Following stroke, microglial cells act in response to injury to scavenge necrotic debris in the CNS [136]. This occurs from the release of pro-inflammatory cytokines almost immediately [112] and activated glial cells, which sense and respond to ATP gradients, act to scavenge excess neurotransmittors and dead cells [137]. The increase in astrocyte activation and proliferation produces the formation of a glial scar, isolating the damaged area from the rest of the CNS [118]. Acute inflammation is advantageous due to the short lived nature and minimization of neuronal damage and toxicity resulting from the release of inflammatory cytokines and chemokines [138]. In a mouse genetic model for Alzheimer’s disease, it has been shown that neurotoxicity of β-Amyloid deposits is reduced through bone marrow derived microglia activation [139]. In addition, Nitric Oxide-induced neuronal damage is increased in mice deficient for TNF-α and TNF-α is required for microglia activation following injury [140]. In addition, in mice IL-6 is important for glial cell activation and is important for neuronal protection following injury to the CNS [141]. Following spinal cord injury, it has been shown that the injection of microglial cells into the site of injury is associated with the regrowth of axons through immune cytochemical detection [142]. In a double blind study with patients with MS, blocking of TNF-α produces an exacerbation of the condition [143]. These studies indicate that cytokine production and glial cell activation and proliferation following injury can act beneficially. However, dysfunctional regulation of neuroinflammation can can also be detrimental. The chronic activation of glial cells and elevation of pro-inflammatory cytokines have been linked to multiple disorders [144]. The development of neuroinflammation can occur multiples ways with different disease etiologies. The inflammatory response can be induced from infection, injury, or an autoimmune disorder. Following the infection of human immunodeficiency virus type 1 (HIV), the disease progresses to Acquired Immune Deficiency Syndrome (AIDS) with the late onset of neurological dysfunction termed AIDS dementia complex (ADC) [145]. ADC is shown to affect roughly 80% of individuals who progress from HIV to AIDS [146]. The progression to ADC has similar mechanisms of other neurological disease through means of neuroinflammation mainly from the activation of glial cells in the CNS [147]. Microglial cells are the primary targets of HIV infection [148] and increases in microglial cells appear to associate with increased viral infection. Histological studies in the CNS from individuals with AIDS show an increase in the proliferation and activation of astrocytes and microglial cells leads to the increased release of IL-1β, TNF-α, and IL-6, which promotes the activation and proliferation of the virus [149]. Through in vitro HIV exposure to human and rat astrocytes, it has been determined that astrocytes are involved through voltage gate and NMDA calcium channels in the progression to ADC [150]. It has further been shown that following ADC, nonviral cells produce proinflammatory cytokines in addition to the viral production of cells in the CNS [151]. GSK3β has been studied in the context of ADC. Through genome wide mRNA and microRNA expression profiles of HIV patients with and without dementia, GSK3β expression is down regulated in patients with dementia [152]. Lithium treatment of mice, in vivo, has been shown to protect the hippocampus from HIV induced neurotoxicity and in vitro lithium exposure prior to infection has shown to reduce HIV induced neurotoxicity [153]. In addition, low dose administration of lithium has been shown to improve the psychological impairment produced from HIV progression [154]. Injury, another inducer of neuroinflammation, can produce strokes (i.e., CNS ischemia), which is a common type of acute brain injury caused by an interruption in the blood supply to the brain where intracellular mechanisms such as inflammation can cause neuronal damage [155]. After the initial injury, macrophages infiltrate the BBB causing an additional inflammatory response to occur [113]. The inflammatory response causes the proliferation and activation of glial cells in the CNS leading to the production of cytokines [136]. Following a stroke, elevated levels of IL-6 and TNF-α in blood or cerebrospinal fluid occur with early clinical symptoms of deterioration [156]. In addition, it has been shown in stroke patients that ischemia causes an abundance of the proinflammatory cytokines IL-6 and TNF-α in peripheral blood cells [157]. Stroke patients have an elevated intrathecal production of proinflammatory cytokines and chemokines indicating there is an increase in glial cell activation [158]. GSK3β has also been studied in the context of stroke. In vivo, following focal cerebral ischemia, GSK3β inhibition has been shown to reduce infarct size [159]. In a rat transient middle cerebral artery occlusion model, pretreatment with lithium decreases infarct size and improves recovery through the increased migration of mesenchymal stem cells [160]. The mechanism by which lithium is beneficial to the treatment of stroke has been thought to occur through activation of the AKT pathway and subsequent phosphorylation of GSK3β, resulting in the attenuation of glutamate excitotoxicity [97]. In addition, pretreatment with lithium has shown to attenuate the dephosphorylation of GSK3β induced through hypoxia in vivo [161]. Multiple Sclerosis (MS) is an autoimmune disorder of the CNS that leads to neurological disability due to axonal deterioration and the pathogenesis of the disorder has been linked to inflammatory elements of MS plaques [162,163]. Briefly, leukocytes infiltrate the CNS through the BBB causing the migration of microglial cells leading towards axonal dysfunction [164]. In post-mortem human brain samples of patients with MS, it has been shown that the cytokines and chemokines RANTES, MCP-1, MIP-1α, and MIP1-β are differentially expressed in glial cells to increase inflammation [165]. TNF has also been shown to be in the lesions of patients with MS indicating an increase of proinflammatory cytokines [166]. Inflammation of the CNS causes the activation of astrocytes and microglial cells which have been targeted in MS. In Act1 deficient mice, it has been shown that astrocytes are necessary for leukocyte recruitment in the CNS in an autoimmune encephalomyelitis model, a model of MS [167]. Inflammatory genes have also been shown to be up regulated in MS plaques [168]. MS is different from other neuroinflammatory mediated diseases in that it originates from immune dysregulation compared to insult/injury of the CNS. GSK3β has also been studied in the context of MS. In experimental autoimmune encephalomyelitis (EAE), over expression of GSK3β increases disease severity, and lithium pretreatment suppresses clinical symptoms and microglial activation in the spinal cord and post treatment promoted partial recovery [169]. In individuals with chronic progressive MS, GSK3β has been shown to be over expressed [170]. Interleukin-17 producing cells, which have been shown to be increased in MS, are inhibited following GSK3 inhibition in an EAE model in the spinal cord [171]. These studies provide evidence that GSK3β is important in disease progression and inhibition of the kinase may play a role in a potential therapeutic. Alzheimer’s disease (AD) is a neurodegenerative disease involving inflammation of the CNS leading to memory loss followed by dementia. It has been suggested that the maincause of AD is due to an abundance of β-Amyloid in the brain [172]. β-Amyloid plaques have been shown to increase the inflammatory response in the brain of Alzheimer’s patients. Glial cells are known to surround Alzheimer’s plaques and in vitro it has been shown that they release IL-1 and growthfactors which are components of plaques [173,174]. In addition, astrocytes have also been shown to surround neuritic plaques in AD [175]. In an age-matched study, Alzheimer’s patients have higher levels of TNF-α in sera indicating an inflammatory response [176]. Using a mouse model for AD, it has been shown that β-Amyloid plaques cause an inflammatory response and LPS stimulation further enhances the productionof the pro-inflammatory cytokines IL-1β, TNF-α and monocytechemo attractant protein-1 [177]. GSK3β has been extensively studied in the context of Alzheimer’s disease. AD patients have been shown to have anincrease in GSK3β activity that correlates with an increase in neuronal death [178]. It has been shown, in vitro, that GSK3β inhibition with lithium treatment blocks the production of β-Amyloid [179]. In cultured rat cortical neurons, lithium protectscells from β-Amyloid induced death and there is a decrease in tau phosphorylation, a process that occurs in response to β-Amyloid accumilation [180]. This was further validated with chronic lithium pretreatment on β-Amyloid induced cerebellar cell death [181]. Clinical studies have also been conducted to determine the benefits of lithium treatment in patients with AD. In elderly patients with both AD and bipolar disorder, chronic lithium treatment has shown to decrease AD symptoms to that seen in the general population [182]. Furthermore, it has been shown that chronic lithium treatment reduces the rate of dementia in a nationwide study [183]. These studies indicate the importance of GSK3β in cognition and neuroinflammation.Neuroinflammation and Pathological Pain
Glia activation and the subsequent release of proinflammatory cytokines play crucial roles in the development and maintenance of pathological pain [184-186]. Microglia and astrocytes are reactivated in almost every animal model of pathological pain [187,188], including neuropathic pain induced by nerve injury [189,190], inflammation induced by complete Freund’s adjuvant [185], surgical incision [191], and morphine tolerance [192]. These are accompanied with elevated levels of proinflammatory cytokines [193,194] and an increased expression of proinflammatory cytokines in microglia and astrocytes in the spinal dorsal horn [195-197], suggesting that the increased proinflammatory cytokines come from glial cells. Intrathecal administration of IL-1β and TNF-α in normal rats enhances both the acute response and the wind-up activity of dorsal horn neurons and mechanical allodynia and hyperalgesia [198,199]. Suppression of astrocyte and microglial activation with the glial inhibitor, propentofylline, or inhibition of microglia activation by minocycline, results in attenuation of hyperalgesia induced by nerve injury, which is associated with decreased expression of the cytokines IL-1β, IL-6 and TNF-α in vivo [194,200,201]. Similarly, treatments with antagonists of IL- 1β, IL-6 and TNF-α reduced hypersensitivity induced by inflammation, nerve injury or morphine tolerance. Besides the release pro-inflammatory cytokines, we and other have shown that activation of astrocytes is associated with dysfunction of glial glutamate transporters [202-204]. Down regulation of glial glutamate transporters in the spinal dorsal horn contributes to the genesis of many types of pathological pain including neuropathic pain induced by nerve injury [202,205,206], chemotherapy [207,208] and morphine tolerance [202,209]. We have demonstrated that impaired glutamate uptake by glial glutamate transporters is a key contributing factor to strengthening AMPA and NMDA receptor activation in thespinal sensory neurons [206,210-213]. Deficiency in glial glutamate uptake results in decreases in GABAergic synaptic strength due to impairment in the GABA synthesis through the glutamate-glutamine cycle [214]. Hence, the integrity of glial GTs is critical to maintain synaptic excitatory-inhibitory homeostasis and normal nociception in the spinal dorsal horn. Glial glutamate transporters appear to be regulated by neuroinflammation processes induced by nerve injury. Suppression of glial activation and pro-inflammatory cytokine production with propentofylline or minocycline up-regulates mRNA and protein expression of glial glutamate transporters in the spinal dorsal horn and ameliorates the nerve-injury-induced allodynia [201,204,215,216]. Therefore, identifying molecules that can suppress neuroinflammation has a great potential to open a new door to alleviate pathological pain. Given the fact that the role of GSK3 in neuroinflammatory diseases in the CNS has been extensively studied and neuroinflammation is a crucial mechanism underlying the genesis of pathological pain, it is surprising that there are only a handful of papers reporting the effects of pharmacological inhibition of GSK3β on spinal nociceptive processing. The first report of GSK3β inhibition on spinal nociceptive processing has been shown in morphine-tolerant rats [217]. Parkitina and coworkers (2006) have shown that GSK3β inhibition alters the tolerance to morphine in the dorsal lumbar of the spinal cord. Intrathecal inhibition of GSK3β (SB216763) has shown to decrease pharmacological tolerance to morphine in a dose-dependent manner whereas GSK3β inhibition of naïve rats has no effect determined from the tailflick test. Tolerance to morphine is associated with an increase in active GSK3β where chronic intrathecal inhibitor administration increases phosphorylation at the Serine-9 residue of GSK3β and decreases tolerance. Intrathecal inhibitor administration has shown to have no effect on increasing the phosphorylation of the Serine-9 residue or have analgesic effects in naïve rats [217]. The next report of GSK3β inhibition on pain processing has been shown in mouse models of acute nociception through acetic acid induced abdominal constrictions and formalin induced nociception [7]. Martins and coworkers (2009) have shown that pharmacological inhibition of GSK3β can have antinociceptive effects. They have shown that intraperitoneal pretreatment of a GSK3β inhibitor (AR-A014418) prior to acetic-acid induced abdominal constrictions reduces writhings. They have also shown that pretreatment with a GSK3β inhibitor by intraperitoneal, intraplantar, and intrathecal injection reduce slicking frequency, a measure of nociception, following formalin induced nociception. Lastly, through intrathecal co-administration of the GSK3β inhibitor with glutamate, NMDA,trans-ACPD, TNF-α, or IL-1β there is a decrease in cytokine induced biting [7]. This study indicates that pharmacological inhibition of GSK3β may play a role in nociception and pain. The most recent report of GSK3β inhibition on pain processing has been shown in a mouse model of neuropathic pain through peripheral nerve injury [5]. Martins and coworkers (2012) have shown that intraperitoneal administration of a GSK3β inhibitor (AR-A014418) produces antihyperalgesic effects and decreases the proinflammatory cytokines IL-1β and TNF-α in the lumbar portion of the spinalcord (Lumbar 1 to Lumbar 6). They have also shown that following the development of mechanical hyperalgesia, a single intraperitoneal administration of a GSK3β inhibitor attenuates mechanical and thermal hyperalgesia. In addition, they have shown that chronic inhibition of serotonin synthesis prior to GSK3β inhibition prevents the decrease in mechanical allodynia. While pharmacological inhibition of GSK3β has been shown to reduce hyperalgesia induced by nerve injury [5], it remains unknown if inhibition of GSK3β can prevent the development of allodynia following nerve injury. In an adult rat (Sprague Dawley) model of neuropathic pain induced by partial sciatic nerve ligation [206], with the same inhibitor (AR-A014418) and concentration (0.3 mg/kg), we found that chronic intraperitoneal administration from the day of surgery (1 injection/day) for 8 days significantly attenuates the development of mechanical allodynia induced by partial sciatic nerve ligation (Figure 5). These data suggest that altered GSK3β activities may contribute to the development of neuropathic pain.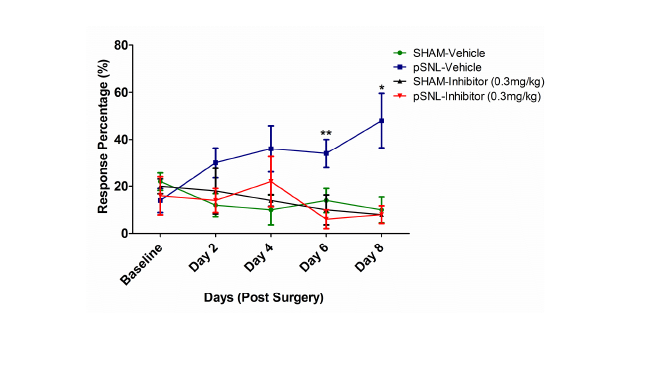
Figure 5: Chronic intraperitoneal injections (IP) of a GSK3β inhibitor prevent the development of allodynia in a model of neuropathic pain. Rats were injected with the GSK3β inhibitor, AR-014418 (0.3 mg/kg), or vehicle (saline) 1 h prior to surgery and then daily for 8 days following surgery. A von Frey filament with 1.5 g force was used to stimulate the plantar region of the hindpaw 10 times and the percentage of hindpaw withdrawal response to 10 time stimulations was used to indicate changes in nociceptive behaviors. Behavior tests were performed before surgery and then prior to the daily drug administration where the examiner was blinded to the types of treatment given to the rats. SHAM rats represent animals where the sciatic nerve was exposed but not ligated. Green represents the SHAM rats receiving vehicle treatment. Blue represents the pSNL group receiving vehicle treatment. Black represents the SHAM rats receiving the GSK3β inhibitor treatment. Red represents the pSNL rats receiving the GSK3β inhibitor. The percentage of hindpaw withdrawal response to the von Frey filament stimulation in pSNL rats receiving the GSK3β inhibitor is significantly lower than that in pSNL rats receiving vehicle treatment, indicating that suppressing GSK3β activities prevents the development of allodynia induced by nerve injury. The asterisks denote a significant difference between the AR-014418 treated pSNL group to the vehicle treated pSNL group. (n=5 per group, Unpaired students t-test.*P **P<0.01).
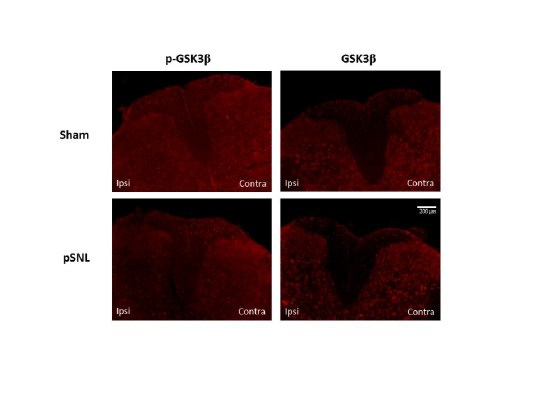
Figure 6: Partial sciatic nerve ligation results in a decrease in phosphorylated GSK3β expression in the injured side of the dorsal horn 8 days following surgery. There was no significant difference in total expression of GSK3β between the pSNL and sham operated rats. Eight days following surgery, rats were deeply anesthetized with pentobarbital (60 mg/kg, IP) and perfused intracardially with 200 mL of heparinized 0.1 M PBS (pH=7.35) followed by 300 mL of a solution containing 4% formaldehyde in 0.1 M PBS (pH=7.35). L4 and L5 spinal segment tissues were then fixed for 48 h at 4°C in the same fixative and cryoprotected for atleast 24 h at 4°C in 30% sucrose in 0.1 M PBS (pH=7.35). Serial transverse sections, 30-μm thick, were cut on a freezing microtome at -20°C and collected in 0.1 M PBS and processed while free floating. The sections were then washed three times in 0.1 M PBS and then blocked with 10% normal goat serum plus 0.3% Triton X-100 in 0.1 M PBS (pH=7.35) for 1 h at room temperature. Sections were then incubated for 24 h at 4°C in rabbit anti-pGSK3β (1:100) or anti-GSK3β (1:200) in 4% normal goat serum plus 0.3% Triton X-100 in 0.1 M PBS (pH=7.35). Sections were then washed 3 times and incubated with with Texas Red goat anti-rabbit antibody (1:500) in 4% normal goat serum plus 0.3% Triton X-100 in 0.1 M PBS (pH=7.35) for 2 h at room temperature. After rinsing in 0.1 M PBS, sections were mounted onto double frosted pre-cleaned microscope slides, air-dried, and cover slipped with UltraCruz Mounting Medium. Slides were imaged using an Olympus 1x71 Inverted Microscope with an Olympus DP72 Microscope Digital Camera. Images were processed using ImageJ (NIH) [218].
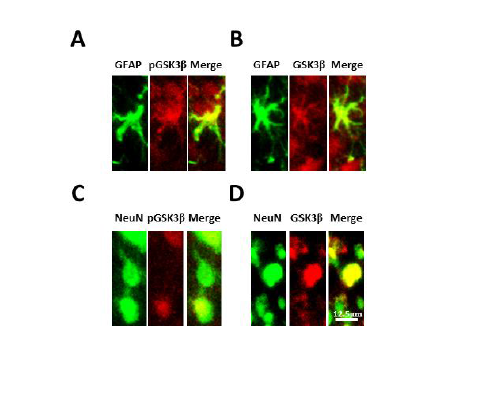
Figure 7: Phosphorylated and total GSK3β are colocalized with neurons and astrocytes in the spinal dorsal horn. GFAP is a marker for astrocytes and NeuN is a marker for neuronal cells. Sections were incubated for 24 h at 4°C in rabbit anti-pGSK3β (1:100) or anti-GSK3β (1:200) in 4% normal goat serum plus 0.3% Triton X-100 in 0.1 M PBS (pH=7.35). Sections were then washed 3 times and incubated with a combination of either mouse anti-NeuN Alexa Fluor 488 conjugated antibody (1:200) or mouse anti-GFAP Alexa Fluor 488 conjugated antibody (1:200) with Texas Red goat anti-rabbit antibody (1:500) in 4% normal goat serum plus 0.3% Triton X-100 in 0.1 M PBS (pH=7.35) for 2 h at room temperature. After rinsing in 0.1 M PBS, sections were mounted, air-dried, cover slipped, and images mentioned in (Figure 6).
Conclusion
Glycogen Synthase Kinase 3β has been linked to the development and progression of multiple disease entities. Following the initial identification of GSK3β, significant strides have been made in understanding the structure, regulation, pharmacology, and diseases linked with the kinase. GSK3β is a common target in inflammation of the CNS which has been associated with many diseases such as Alzheimer’s disease, AIDS dementia complex, and stroke. Inhibition of GSK3β has been shown to alleviate multiple symptoms and the progression of these diseases. The role of GSK3β in pathological pain has recently emerged. The development and maintenance of pathological pain is associated with changes in GSK3β activities. Inhibition of GSK3β activities can prevent the development and reverse the existence of neuropathic pain. Further studies are needed to understand the upstream pathways regulating GSK3β activities and the downstream signaling pathways used by GSK3β to alter spinal sensory processing, which will potentially lead to the use of GSK3β as a novel target for the development of analgesics.Acknowledgments
project was supported by NIH RO1 grant NS064289 to HRW.References
- Hanks SK, Hunter T (1995) Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. The FASEB Journal. 9: 576-596.
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH (1992) Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 147: 58-62.
- Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769-776.
- Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science Signalling 296: 1655-1657.
- Mazzardo ML, Martins DF, Stramosk J, Cidral-Filho FJ, Santos AR (2012) Glycogen Synthase Kinase 3-Specific Inhibitor AR-A014418 Decreases Neuropathic Pain in Mice: Evidence for The Mechanisms of Action. Neuroscience 226: 411-420.
- Jope RS, Yuskaitis CJ, Beurel E (2007) Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res 32: 577-595.
- Martins DF, Rosa AO, Gadotti VM, Mazzardo ML, Nascimento FP (2011) The antinociceptive effects of AR-A014418, a selective inhibitor of glycogen synthase kinase-3 beta, in mice. The Journal of Pain. 12: 315-322.
- Woodgett JR (1990) Molecular cloning and expression of glycogen synthase kinase-3/Factor A. EMBO J 9: 2431-2438.
- Ferrer I, Barrachina M, Puig B (2002) Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Acta neuropathol 104: 583-591.
- Shiurba RA, Ishiguro K, Takahashi M, Sato K, Spooner ET, et al. (1996) Immunocytochemistry of tau phosphoserine 413 and tau protein kinase I in Alzheimer pathology. Brain Res 737: 119-132.
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, et al. (1992) Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Letters 314: 315-321.
- Hernández F, Pérez M, Lucas JJ, Mata AM, Bhat R, et al. (2004) Glycogen Synthase Kinase-3 Plays a Crucial Role in Tau Exon 10 Splicing and Intranuclear Distribution of SC35: IMPLICATIONS FOR Alzheimer’s DISEASE. J Biol Chem 5: 3801-3806.
- Souza RP, Romano-Silva MA, Lieberman JA, Meltzer HY, Wong AH, et al. (2008) Association study of GSK3 gene polymorphisms with schizophrenia and clozapine response. Psychopharmacology 200: 177-186.
- Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA, et al. (2004) Convergent evidence for impaired AKT1-GSK3β signaling in schizophrenia. Nature Genetics 36: 131-137.
- Peterson JW, Bö L, Mörk S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50: 389-400.
- Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE (1990) Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis Pathogenetic and clinical implications. Brain 113: 1477-1489.
- Thompson KA, McArthur JC, Wesselingh SL (2001) Correlation between neurological progression and astrocyte apoptosis in HIV-associated dementia. Ann Neurol 49: 745-752.
- McArthur JC (2004) HIV dementia: an evolving disease. J Neuroimmunol 157: 3-10.
- Stolp HB, Dziegielewska KM (2009) Review: Role of developmental inflammation and blood–brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol 35: 132-146.
- Noble W, Planel E, Zehr C, Olm V, Meyerson J, et al. (2005) Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 102: 6990-6995.
- Martinez A, Perez DI (2008) GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer’s disease? J Alzheimers Dis. 15: 181-191.
- Martinez A (2008) Preclinical efficacy on GSK-3 inhibitors: Towards a future generation of powerful drugs. Med Res Rev 28: 773-796.
- Lipina TV, Kaidanovich-Beilin O, Patel S, Wang M, Clapcote SJ et al. (2011) Genetic and pharmacological evidence for schizophrenia-related Disc1 interaction with GSK-3. Synapse 65: 234-248.
- Scholz J, Woolf CJ (2002) Can we conquer pain? Nat Neurosci 5: 1062-1067.
- Woolf CJ (1983) Evidence for a central component of post-injury pain hypersensitivity. Nature 306: 686-688.
- Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science. 288: 1765-1768.
- Woolf CJ, Shortland P, Coggeshall RE (1992) Peripheral nerve injury triggers central sprouting of myelinated afferents. Nature 2: 75-78.
- Embi N, Rylatt DB, Cohen P (1980) Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 107: 519-527.
- Friedman DL, Larner J (1963) Studies on UDPG-α-Glucan Transglucosylase. III. Interconversion of Two Forms of Muscle UDPG-α-Glucan Transglucosylase by a Phosphorylation-Dephosphorylation Reaction Sequence. Biochemistry 2: 669-675.
- Schlender KK, Beebe SJ, Willey JC, Lutz SA, Reimann EM (1980) Isolation and characterization of cyclic AMP-independent glycogen synthase kinase from rat skeletal muscle. Biochem Biophys Acta 615: 324-340.
- Cohen P, Yellowlees D, Aitken A, Donella-Deana A, Hemmings BA (1982) Separation and Characterisation of Glycogen Synthase Kinase 3, Glycogen Synthase Kinase 4 and Glycogen Synthase Kinase 5 from Rabbit Skeletal Muscle. Eur J Biochem 124: 21-35.
- Picton C, Aitken A, Bilham T, Cohen P (1982) Multisite Phosphorylation of Glycogen Synthase from Rabbit Skeletal Muscle. Eur J Biochem 124: 37-45.
- Woodgett JR, Cohen P (1984) Multisite phosphorylation of glycogen synthase: Molecular basis for the substrate specificity of glycogen synthease kinase-3 and casein kinase-II (glycogen synthase kinase-5). Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology 788: 339-347.
- Woodgett JR (1991) cDNA cloning and properties of glycogen synthase kinase-3. Methods In Enzymol 200: 564-577.
- Sutherland C, Leighton IA, Cohen P (1993) Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 296: 15-19.
- Woodgett JR (1994) Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol 5: 269-275.
- Farkas IJ, Szántó VÁ, Korcsmáros T (2012) Linking proteins to signaling pathways for experiment design and evaluation. PLoS One 7: e36202.
- Ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, et al. (2001) Structure of GSK3β reveals a primed phosphorylation mechanism. Nat Struct Biology 8: 593-596.
- Bhat R, Xue Y, Berg S, Hellberg S, Ormö M, et al, (2003) Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-specific Inhibitor AR-A014418. J Biol Chem 278: 45937-45945.
- Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Progress in Neurobiology. 65: 391-426.
- Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochemical Journal 359: 1-16.
- Sutherland C, Leighton IA, Cohen P (1993) Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem J 296: 15-19.
- Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, et al. (2008) Phosphorylation by p38 MAPK as an Alternative Pathway for GSK3 {beta} Inactivation. Science Signalling. 320: 667-670.
- Ding Q, Xia W, Liu JC, Yang JY, Lee DF, et al. (2005) Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Molecular cell 19: 159-170.
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR (1993) Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. The EMBO journal. 12: 803-808.
- Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781-810.
- Wu D, Pan W (2010) GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 35: 161-68.
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789.
- Cohen P (1985) The coordinated control of metabolic pathways by broad-specificity protein kinases and phosphatases. Curr Top Cellular Regul 27: 23-37.
- Cohen P (1979) The hormonal control of glycogen metabolism in mammalian muscle by multivalent phosphorylation. Biochem Soc Trans 7: 459-480.
- Parker PJ, Caudwell FB, Cohen P (1983) Glycogen synthase from rabbit skeletal muscle; effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur J Biochem 130: 227-234.
- Hughes K, Ramakrishna S, Benjamin WB, Woodgett JR (1992) Identification of multifunctional ATP-citrate lyase kinase as the α-isoform of glycogen synthase kinase-3. Biochem J 15: 309-314.
- Cross DA, Alessi DR, Vandenheede JR, McDowell HE, Hundal HS, et al. (1994) The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin, but not by rapamycin: evidence that wortmannin blocks activation of the mitogen-activated protein kinase pathway in L6 cells between Ras and Raf. Biochem J 303: 21-26.
- Armstrong JL, Bonavaud SM, Toole BJ, Yeaman SJ (2001) Regulation of Glycogen Synthesis by Amino Acids in Cultured Human Muscle Cells. J Biol Chem 276: 952-956.
- Zhang HH, Lipovsky AI, Dibble CC, Sahin M, Manning BD (2006) S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Molecular Cell 24: 185-197.
- Kim L, Liu J, Kimmel AR (1999) The Novel Tyrosine Kinase ZAK1 Activates GSK3 to Direct Cell Fate Specification. Cell 99: 399-408.
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, et al. (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci 97: 11074-11079.
- Sayas CL, Moreno-Flores MT, Avila J, Wandosell F (1999) The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J Biol Chem 274: 37046-37052.
- Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH (2006) GSK-3 is activated by the tyrosine kinase Pyk2 during LPA1-mediated neurite retraction. Mol Biol Cell 17: 1834-1844.
- Salic A, Lee E, Mayer L, Kirschner MW (2000) Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell 5: 523-532.
- Jia J, Amanai K, Wang G, Tang J, Wang B, et al. (2002) Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416: 548-552.
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, et al. (1998) Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3[beta] and [beta]-catenin and promotes GSK-3[beta]-dependent phosphorylation of [beta]-catenin. EMBO J 17: 1371-1384.
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, et al, (1997) The Mouse Fused Locus Encodes Axin, an Inhibitor of the Wnt Signaling Pathway That Regulates Embryonic Axis Formation. Cell 90: 181-192.
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, et al. (1999) Phosphorylation of Axin, a Wnt Signal Negative Regulator, by Glycogen Synthase Kinase-3β Regulates Its Stability. J Biol Chem 274: 10681-10684.
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, et al. (2002) Axin-mediated CKI phosphorylation of β-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16: 1066-1076.
- Kimelman D, Xu W (2006) β-Catenin destruction complex: insights and questions from a structural perspective. Oncogene 25: 7482-7491.
- Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, et al. (1999) An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J 18: 2401-2410.
- Polakis P (2000) Wnt signaling and cancer. Genes Dev 14: 1837-1851.
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P (1998) Downregulation of β-catenin by human Axin and its association with the APC tumor suppressor, β-catenin and GSK3. Curr Biol 8: 573-581.
- Metcalfe C, Bienz M (2011) Inhibition of GSK3 by Wnt signalling–two contrasting models. J Cell Sci 124: 3537-3544.
- Fan CM, Porter JA, Chiang C, Chang DT, Beachy PA, et al. (1995) Long-range sclerotome induction by sonic hedgehog: Direct role of the amino-terminal cleavage product and modulation by the cyclic AMP signaling pathway. Cell 81: 457-465.
- Aikin RA, Ayers KL, Thérond PP (2008) The role of kinases in the Hedgehog signalling pathway. EMBO Rep 9: 330-336.
- Chen Y, Gallaher N, Goodman RH, Smolik SM (1998) Protein kinase A directly regulates the activity and proteolysis of cubitus interruptus. Proc Natl Acad Sci 95: 2349-2354.
- Hernández F, Nido JD, Avila J, Villanueva N (2009) GSK3 inhibitors and disease. Mini Rev Med Chem 9: 1024-1029.
- Beurel E (2011) Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front Mol Neurosci 4: 18.
- Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, et al. (2005) GSK3B polymorphisms alter transcription and splicing in Parkinson's disease. Ann Neurol 58: 829-839.
- Eldar-Finkelman H (2002) Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med 8: 126-132.
- Martinez A, Castro A, Dorronsoro I, Alonso M (2002) Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med Res Rev 22: 373-384.
- Rowe MK, Wiest C, Chuang DM (2007) GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci Biobehav Rev 31: 920-931.
- Forde JE, Dale TC (2007) Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 64: 1930-1944.
- ClinicalTrials gov (2009) Safety Study of a Glycogen Synthase Kinase 3 (GSK3) Inhibitor in Patients With Alzheimer’s Disease.
- ClinicalTrials gov (2012) The Efficacy and Safety of Topical Valproic Acid in Preventing Hair Loss.
- ClinicalTrials gov (2012) Trial of Valproic Acid in Patients With Progressive Supranuclear Palsy (Depakine).
- Clinical Trials gov (2012) Safety, Tolerability, and Efficacy of Two Different Oral Doses of NP031112 Versus Placebo in the Treatment of Patients With Mild-to-Moderate Progressive Supranuclear Palsy (Tauros).
- ClinicalTrials gov (2009) Investigation of Lithium on Signal Transduction, Gene Expression and Brain Myo-Inositol Levels in Manic Patients.
- Clinical Trials gov (2012) Simvastatin Augmentation of Lithium Treatment in Bipolar Depression.
- ClinicalTrials gov (2012) Neural Correlates for Therapeutic Mechanisms of Lithium in Bipolar Disorder.
- ClinicalTrials gov (2012) Characterization of the Changes in the Signalling Pathways During Spinal Cord Injury-induced Skeletal Muscle Atrophy.
- ClinicalTrials gov (2012) A Phase I Trial of Enzastaurin (LY317615) in Combination With Carboplatin in Adults With Recurrent Gliomas.
- Klein PS, Melton DA (1996) A Molecular Mechanism for the Effect of Lithium on Development. Proc Natl Acad Sci U S A 93: 8455-8459.
- Bowden CL, Calabrese JR, McElroy SL, Gyulai L, Wassef A, et al. (2000) A randomized, placebo-controlled 12-month trial of divalproex and lithium in treatment of outpatients with bipolar I disorder. Arch Gen Psychiatry 57: 481-489.
- Stambolic V, Ruel L, Woodgett JR (1996) Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr Biol 6: 1664-1669.
- Takahashi M, Yasutake K, Tomizawa K (1999) Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J Neurochem 73: 2073-2083.
- Ryves WJ, Harwood AJ (2001) Lithium Inhibits Glycogen Synthase Kinase-3 by Competition for Magnesium. Biochem Biophys Res Commun 280: 720-725.
- Ryves WJ, Dajani R, Pearl L, Harwood AJ (2002) Glycogen synthase kinase-3 inhibition by lithium and beryllium suggests the presence of two magnesium binding sites. Biochem Biophys Res Commun 290: 967-972.
- Phiel CJ, Klein PS (2001) Molecular targets of lithium action. Ann Rev Pharmacol Toxicol 41: 789-813.
- Chalecka-Franaszek E, Chuang DM (1999) Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci 96: 8745-8750.
- Jacobs KM, Bhave SR, Ferraro DJ, Jaboin JJ, Hallahan DE, et al. (2012) GSK-3: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol 2012:930710.
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, et al. (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7: 793-803.
- Martin M, Rehani K, Jope RS, Michalek SM (2005) Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 6: 777-784.
- Götschel F, Kern C, Lang S, Sparna T, Markmann C, et al. (2008) Inhibition of GSK3 differentially modulates NF-κB, CREB, AP-1 and β-catenin signaling in hepatocytes, but fails to promote TNF-α-induced apoptosis. Exp Cell Res 314: 1351-1366.
- Grimes CA, Jope RS (2001) CREB DNA binding activity is inhibited by glycogen synthase kinase-3β and facilitated by lithium. J Neurochem 78: 1219-1232.
- Wen AY, Sakamoto KM, Miller LS (2010) The Role of the Transcription Factor CREB in Immune Function. J Immunol 85: 6413-6419.
- Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, et al. (2007) GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood 109: 1584-1592.
- Hofmann C, Dunger N, Schölmerich J, Falk W, Obermeier F (2010) Glycogen synthase kinase 3-β: A master regulator of toll-like receptor-mediated chronic intestinal inflammation. Inflamm Bowel Dis 16: 1850-1858.
- Tak PP, Firestein GS (2001) Firestein, NF-kappaB: a key role in inflammatory diseases. J Clin Invest 107: 7-11.
- Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA (2001) Possible new role for NF-[kappa]B in the resolution of inflammation. Nat Med 7: 1291-1297.
- Foxwell B, Browne K, Bondeson J, Clarke C, de Martin R, et al. (1998) Efficient adenoviral infection with IκBα reveals that macrophage tumor necrosis factor α production in rheumatoid arthritis is NF-κB dependent. Proc Natl Acad Sci USA 95: 8211-8215.
- Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ (2004) Molecular mechanisms of interleukin-10-mediated inhibition of NF-κB activity: a role for p50. Clin Exp Immunol 135: 64-73.
- Antoniv TT, Ivashkiv LB (2011) Interleukin-10-induced gene expression and suppressive function are selectively modulated by the PI3K-Akt-GSK3 pathway. Immunology 132: 567-577.
- Schwabe RF, Brenner DA (2002) Role of glycogen synthase kinase-3 in TNF-α-induced NF-κB activation and apoptosis in hepatocytes. Am J Physiol Gastrointest Liver Physiol 283: G204-G211.
- Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:14.
- de Vries HE, Kuiper J, de Boer AG, Van Berkel TJ, Breimer DD (1997) The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev 49: 143-155.
- Green HF, Nolan YM (2012) GSK-3 mediates the release of IL-1β, TNF-α and IL-10 from cortical glia. Neurochem Int 61: 666-671.
- Beurel E, Michalek SM, Jope RS (2010) Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 31: 24-31.
- Lampson LA (1987) Molecular bases of the immune response to neural antigens. Trends in Neurosciences 10: 211-216.
- Roberts TK, Buckner CM, Berman JW (2010) Leukocyte transmigration across the blood-brain barrier: perspectives on neuroAIDS. Front Biosci 15: 478-536.
- Lucas SM, Rothwell NJ, Gibson RM (2006) The role of inflammation in CNS injury and disease. Br J Pharmacol 147 Suppl 1: S232-S240.
- Consilvio C, Vincent AM, Feldman EL (2004) Neuroinflammation, COX-2, and ALS—a dual role? Exp Neurol 187: 1-10.
- Moalem G, Tracey DJ (2006) Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev 51: 240-264.
- del Rio-Hortega P (1993) Art and artifice in the science of histology. Histopathology 22: 515-525.
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314-1318.
- Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9: 429-39.
- Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC (2006) CNS immune privilege: hiding in plain sight. Immunol Rev 213: 48-65.
- Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8: 57-69.
- Kielian T (2004) Microglia and chemokines in infectious diseases of the nervous system: views and reviews. Front Biosci 9: 732-750.
- Cartier L, Hartley O, Dubois-Dauphin M, Krause KH (2005) Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev 48: 16-42.
- Maragakis NJ, Rothstein JD (2006) Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2: 679-689.
- Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1-14.
- Liberto CM, Albrecht PJ, Herx LM, Yong VW, Levison SW (2004) Pro-regenerative properties of cytokine-activated astrocytes. J Neurochem 89: 1092-1100.
- Ridet JL, Malhotra SK, Privat A, Gage FH (1997) Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci 20: 570-577.
- Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22: 183-192.
- Lau LT, Yu AC (2001) Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma 18: 351-359.
- Kim SU, de Vellis J (2005) Microglia in health and disease. J Neurosci Res 81:302-313.
- Lieberman AP, Pitha PM, Shin HS, Shin ML (1989) Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad Sci USA 86:6348-6352.
- Danton GH, Dietrich WD (2003) Inflammatory Mechanisms after Ischemia and Stroke. J Neuropathol Exp Neurol 62: 127-136.
- Popovich PG, Longbrake EE (2008) Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci 9: 481-493.
- Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Does neuroinflammation fan the flame in neurodegenerative diseases. Mol Neurodegener 4: 47.
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron 49: 489-502.
- Turrin NP, Rivest S (2006) Tumor necrosis factor α but not interleukin 1β mediates neuroprotection in response to acute nitric oxide excitotoxicity. J Neurosci 26: 143-151.
- Penkowa M, Moos T, Carrasco J, Hadberg H, Molinero A, et al. (1999) Strongly compromised inflammatory response to brain injury in interleukin-6-deficient mice. Glia 25: 343-357.
- Prewitt CM, Niesman IR, Kane CJ, Houlé JD (1997) Activated Macrophage/Microglial Cells Can Promote the Regeneration of Sensory Axons into the Injured Spinal Cord. Exp Neurol 148: 433-443.
- Group, L.M.S.S. (1999) TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. University of British Columbia MS/MRI Analysis Group. Neurology 53: 457-465.
- Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14: 1189-1197.
- Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, et al. (1988) The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science 239: 586-592.
- Spencer DC, Price RW (1992) Human immunodeficiency virus and the central nervous system. Annu Rev Microbiol 46: 655-693.
- Watkins BA, Dorn HH, Kelly WB, Armstrong RC, Potts BJ, et al. (1990) Specific tropism of HIV-1 for microglial cells in primary human brain cultures. Science 249: 549-553.
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7: 75-83.
- Merrill JE, Chen IS (1991) HIV-1, macrophages, glial cells, and cytokines in AIDS nervous system disease. FASEB J 5: 2391-2397.
- Benos DJ, Hahn BH, Bubien JK, Ghosh SK, Mashburn NA, et al. (1994) Envelope glycoprotein gp120 of human immunodeficiency virus type 1 alters ion transport in astrocytes: implications for AIDS dementia complex. Proc Natl Acad Sci USA 91: 494-498.
- Nuovo GJ, Alfieri ML (1996) AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med 2: 358-366.
- Zhou L, Pupo GM, Gupta P, Liu B, Tran SL, et al. (2012) A parallel genome-wide mrna and microrna profiling of the frontal cortex of HIV patients with and without HIV-associated dementia shows the role of axon guidance and downstream pathways in HIV-mediated neurodegeneration. BMC Genomics 13: 677.
- Everall IP, Bell C, Mallory M, Langford D, Adame A, et al. (2002) Lithium Ameliorates HIV-gp120-Mediated Neurotoxicity. Mol Cell Neurosci 21: 493-501.
- Letendre SL, Woods SP, Ellis RJ, Atkinson JH, Masliah E, et al. (2006) Lithium improves HIV-associated neurocognitive impairment. AIDS 20: 1885-1888.
- White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, et al. (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179: 1-33.
- Vila N, Castillo J, Dávalos A, Chamorro A (2000) Proinflammatory Cytokines and Early Neurological Worsening in Ischemic Stroke. Stroke 31: 2325-2329.
- Ferrarese C, Mascarucci P, Zoia C, Cavarretta R, Frigo M, et al. (1999) Increased Cytokine Release From Peripheral Blood Cells After Acute Stroke. J Cereb Blood Flow Metab 19: 1004-1009.
- Tarkowski E, Rosengren L, Blomstrand C, Wikkelsö C, Jensen C, et al. (1997) Intrathecal release of pro- and anti-inflammatory cytokines during stroke. Clin Exp Immunol 110: 492-499.
- Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, et al. (2004) Glycogen synthase kinase 3β inhibitor Chir025 reduces neuronal death resulting from oxygen-glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol 188: 378-386.
- Tsai LK, Wang Z, Munasinghe J, Leng Y, Leeds P, et al. (2011) Mesenchymal Stem Cells Primed With Valproate and Lithium Robustly Migrate to Infarcted Regions and Facilitate Recovery in a Stroke Model. Stroke 42: 2932-2939.
- Roh MS, Eom TY, Zmijewska AA, De Sarno P, Roth KA, et al. (2005) Hypoxia activates glycogen synthase kinase-3 in mouse brain in vivo: Protection by mood stabilizers and imipramine. Biol Psychiatry 57: 278-286.
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, et al. (1998) Axonal Transection in the Lesions of Multiple Sclerosis. N Engl J Med 338: 278-285.
- Frohman EM, Racke MK, Raine CS (2006) Multiple Sclerosis — The Plaque and Its Pathogenesis. N Engl J Med 354: 942-955.
- Merrill JE, Benveniste EN (1996) Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci 19: 331-338.
- Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN (1988) Expression of monocyte chemoattractant protein-1 and other β-chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J Neuroimmunol 84: 238-249.
- Hofman FM, Hinton DR, Johnson K, Merrill JE (1989) Tumor necrosis factor identified in multiple sclerosis brain. J Exp Med 170: 607-612.
- Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, et al. (2010) Astrocyte-Restricted Ablation of Interleukin-17-Induced Act1-Mediated Signaling Ameliorates Autoimmune Encephalomyelitis. Immunity 32: 414-425.
- Mycko MP, Papoian R, Boschert U, Raine CS, Selmaj KW (2003) cDNA microarray analysis in multiple sclerosis lesions: detection of genes associated with disease activity. Brain 126: 1048-1057.
- De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, et al. (2008) Lithium Prevents and Ameliorates Experimental Autoimmune Encephalomyelitis. J Immunol 181: 338-345.
- Booth DR, Arthur AT, Teutsch SM, Bye C, Rubio J, et al. (2005) Gene expression and genotyping studies implicate the interleukin 7 receptor in the pathogenesis of primary progressive multiple sclerosis. J Mol Med (Berl) 83: 822-830.
- Beurel E, Yeh WI, Michalek SM, Harrington LE, Jope RS (2011) Glycogen Synthase Kinase-3 Is an Early Determinant in the Differentiation of Pathogenic Th17 Cells. J Immunol 186: 1391-1398.
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297: 353-356.
- Araujo DM, Cotman CW (1992) β-Amyloid stimulates glial cells in vitro to produce growth factors that accumulate in senile plaques in Alzheimer's disease. Brain Res 569: 141-145.
- Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, Lossinsky AS (1990) Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl 7: 287-297.
- Mrak RE, Sheng JG, Griffin WS (1995) Glial cytokines in Alzheimer’s disease: review and pathogenic implications. Hum Pathol 26: 816-823.
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, et al. (1991) Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett 129: 318-320.
- Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, et al. (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull 56: 581-588.
- Bhat RV, Budd Haeberlein SL, Avila J (2004) Glycogen synthase kinase 3: a drug target for CNS therapies. J Neurochem 89: 1313-1317.
- Sun X, Sato S, Murayama O, Murayama M, Park JM, et al. (2002) Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci Lett 321: 61-64.
- Alvarez G, Muñoz-Montaño JR, Satrústegui J, Avila J, Bogónez E, et al. (1999) Lithium protects cultured neurons against β-amyloid-induced neurodegeneration. FEBS Lett 453: 260-264.
- Wei H, Leeds PR, Qian Y, Wei W, Chen R, et al. (2000) β-Amyloid peptide-induced death of PC 12 cells and cerebellar granule cell neurons is inhibited by long-term lithium treatment. Eur J Pharmacol 392: 117-123.
- Nunes PV, Forlenza OV, Gattaz WF (2007) Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry 190: 359-360.
- Kessing LV, Søndergård L, Forman JL, Andersen PK (2008) Lithium treatment and risk of dementia. Arch Gen Psychiatry 65: 1331-1335.
- Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10: 23-36.
- Ren K, Dubner R (2010) Interactions between the immune and nervous systems in pain. Nat Med 16: 1267-1276.
- Gao YJ, Ji RR (2010) Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther 126: 56-68.
- Wieseler-Frank J, Maier SF, Watkins LR (2005) Central proinflammatory cytokines and pain enhancement. Neurosignals 14: 166-174.
- Wieseler-Frank J, Maier SF, Watkins LR (2005) Immune-to-brain communication dynamically modulates pain: physiological and pathological consequences. Brain Behav Immun 19: 104-111.
- Coyle DE (1998) Partial peripheral nerve injury leads to activation of astroglia and microglia which parallels the development of allodynic behavior. Glia 23: 75-83.
- Honore P, Rogers SD, Schwei MJ, Salak-Johnson JL, Luger NM, et al. (2000) Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience 98: 585-598.
- Peters CM, Eisenach JC (2010) Contribution of the Chemokine CCL2 to Mechanical Hypersensitivity Following Surgical Incision in Rats. Anesthesiology 112: 1250.
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF (2005) Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 28: 661-669.
- DeLeo JA, Colburn RW, Nichols M, Malhotra A (1996) Interleukin-6-mediated hyperalgesia/allodynia and increased spinal IL-6 expression in a rat mononeuropathy model. J Interferon Cytokine Res 16: 695-700.
- DeLeo, JA, Colburn RW, Rickman AJ (1997) Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Research 759: 50-57.
- Schäfers M, Svensson CI, Sommer C, Sorkin LS (2003) Tumor necrosis factor-α induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 23: 2517-2521.
- Ohtori S, Takahashi K, Moriya H, Myers RR (2004) TNF-α and TNF-α receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine 29: 1082-1028.
- George A, Buehl A, Sommer C (2005) Tumor necrosis factor receptor 1 and 2 proteins are differentially regulated during Wallerian degeneration of mouse sciatic nerve. Exp Neurol 192: 163-166.
- Reeve AJ, Patel S, Fox A, Walker K, Urban L (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 4: 247-257.
- Kwon BK, Fisher CG, Dvorak MF, Tetzlaff W (2005) Strategies to promote neural repair and regeneration after spinal cord injury. Spine 30: S3-S13.
- Raghavendra V, Tanga F, DeLeo JA (2003) Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 306: 624-630.
- Sweitzer SM, Schubert P, DeLeo JA (2001) Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol ExpTher 297: 1210-1217.
- Ramos KM, Lewis MT, Morgan KN, Crysdale NY, Kroll JL, et al. (2010) Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience 169: 1888-1900.
- Nie H, Zhang H, Weng HR (2010) Minocycline prevents impaired glial glutamate uptake in the spinal sensory synapses of neuropathic rats. Neuroscience 170: 901-912.
- Tawfik VL, Regan MR, Haenggeli C, Lacroix-Fralish ML, Nutile-McMenemy N, et al., (2008) Propentofylline-induced astrocyte modulation leads to alterations in glial glutamate promoter activation following spinal nerve transection. Neurosci 152: 1086-1092.
- Sung B, Lim G, Mao J (2003) Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci 23: 2899-2910.
- Nie H, Weng HR (2010) Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J Neurophysiol 103: 2570-2580.
- Weng HR, Aravindan N, Cata JP, Chen JH, Shaw AD, et al. (2005) Spinal glial glutamate transporters downregulate in rats with taxol-induced hyperalgesia. Neurosci Lett 386: 18-22.
- Doyle T, Chen Z, Muscoli C, Bryant L, Esposito E, Cuzzocrea S, et al. (2012) Targeting the Overproduction of Peroxynitrite for the Prevention and Reversal of Paclitaxel-Induced Neuropathic Pain. J Neurosci 32: 6149-6160.
- Mao J, Sung B, Ji RR, Lim G (2002) Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312-8323.
- Weng H, Chen J, Cata J (2006) Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience 138: 1351-1360.
- Weng HR, Chen JH, Pan ZZ, Nie H (2007) Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience 149: 898-907.
- Nie H, Weng HR (2009) Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J Neurophysiol 101: 2041-2051.
- Nie H, Zhang H, Weng HR (2010) Bidirectional Neuron–Glia Interactions Triggered by Deficiency of Glutamate Uptake at Spinal Sensory Synapses. J Neurophysiol 10: 713-725.
- Jiang E, Yan X, Weng HR (2012) Glial glutamate transporter and glutamine synthetase regulate GABAergic synaptic strength in the spinal dorsal horn. J neurochem 12: 526-536.
- Tawfik VL, Lacroix-Fralish ML, Bercury KK, Nutile-McMenemy N, Harris BT, et al. (2006) Induction of astrocyte differentiation by propentofylline increases glutamate transporter expression in vitro: heterogeneity of the quiescent phenotype. Glia 54: 193-203.
- Nie H, Zhang H, Weng HR (2010) Minocycline prevents impaired glial glutamate uptake in the spinal sensory synapses of neuropathic rats. Neuroscience. 170: 901-912.
- Parkitna JR, Obara I, Wawrzczak-Bargiela A, Makuch W, Przewlocka B, et al. (2006) Effects of Glycogen Synthase Kinase 3β and Cyclin-Dependent Kinase 5 Inhibitors on Morphine-Induced Analgesia and Tolerance in Rats. J Pharmacol Exp Ther 319: 832-839.
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671-675.