
Journal of Pharmaceutics & Pharmacology
Download PDF
Review Article
*Address for Correspondence: Jun-Feng Wang, Departments of Pharmacology and Therapeutics, and Psychiatry, University of Manitoba, Winnipeg, Canada, E-mail: Jun-Feng.Wang@med.umanitoba.ca
Citation: Wang Y, Wang JF. The Role of Protein S-Nitrosylationin Alzheimer’s Disease and its Treatment. J Pharmaceu Pharmacol. 2015;3(1): 6.
Copyright © 2015 Wang Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology |ISSN: 2327-204X| Volume: 3, Issue: 1
Submission: 02 February 2015 | Accepted: 07 March 2015 | Published: : 12 March 2015
Although the mechanisms of AD are not fully understood, Aβ plaque, neurofibrillary tangles and neurotransmission deficits have been identified to be its main characteristics [7-9]. Aβ plaque resultsfrom aggregation of Aβ peptides that originate from cleavage of APP by β-secretase and γ-secretase [7]. It has been found that Aβ oligomers are toxic, and can cause synapse loss and neuronal cell death [10]. Neurofibrillary tangles are aggregates of hyperphosphorylated tau proteins in the brain [11]. Tau protein is expressed in axons and binds to tubulin, which stabilizes microtubules. In AD patients,hyperphosphorylated tau protein aggregates into insoluble tangles, which have a detrimental effect on the transport system of neurons,resulting in disconnection of neurons and eventually cell death [8,9]. Since AD is an age-dependent disease that mainly occurs in the elderly, ageing is a leading factor. The free radicals that can cause oxidative/nitrosative stress contribute significantly to the aging process. Indeed, many studies have reported that the AD brain exhibits increased oxidative/nitrosative stress [12]. Increasing evidence suggests that protein S-nitrosylation plays an important role in the pathophysiological development of AD [13,14].

Protein misfolding and aggregation are increased in brain of patients with AD. For instance, neurofibrillary tangles are caused by hyperphosphorylated tau aggregates, while Aβ plaques are caused by Aβ aggregates [7-9]. Recent studies show that S-nitrosylation of protein disulfide isomerase (PDI) in AD contributes to misfolding of proteins. The immature proteins are synthesized on ribosomes, and then translocated to endoplasmic reticulum for proper folding by disulfide bond formation, which makes proteins mature and ready for functioning. PDI catalyzes the correct formation of disulfide bond based on thiol-disulfide exchange reactions [53,57]. PDI also serves as a molecular chaperone that assists in the maturation, transport and folding of secretary proteins [40]. It is evidenced that the absence of proper disulfide bond formation in the endoplasmic reticulum is closely related to down regulation of PDI enzyme activity. Low protein activity of PDI can cause endoplasmic reticulum stress, which is a conventional signal pathway for apoptosis [55]. Therefore PDI can reduce protein misfolding and attenuate endoplasmic reticulum stress, subsequently preventing neuronal cell death.
The Role of Protein S-Nitrosylationin Alzheimer’s Disease and its Treatment
Ying Wang1,2 and Jun-Feng Wang1,2*
- 1Kleysen Institute for Advanced Medicine, Winnipeg Health Sciences Centre, Canada
- 2Departments of Pharmacology and Therapeutics, and Psychiatry, University of Manitoba, Winnipeg, Canada
*Address for Correspondence: Jun-Feng Wang, Departments of Pharmacology and Therapeutics, and Psychiatry, University of Manitoba, Winnipeg, Canada, E-mail: Jun-Feng.Wang@med.umanitoba.ca
Citation: Wang Y, Wang JF. The Role of Protein S-Nitrosylationin Alzheimer’s Disease and its Treatment. J Pharmaceu Pharmacol. 2015;3(1): 6.
Copyright © 2015 Wang Y, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Pharmaceutics & Pharmacology |ISSN: 2327-204X| Volume: 3, Issue: 1
Submission: 02 February 2015 | Accepted: 07 March 2015 | Published: : 12 March 2015
Abstract
As a post-transcriptional modification, S-nitrosylation is a covalent binding of nitric oxide to cysteine thiols in proteins to form S-nitrosothiols. Nitric oxide is recognized as an important signaling molecule, and has recently been shown to reversibly nitrosylated proteins. Emerging evidence demonstrates that in Alzheimer’s disease, aberrant S-nitrosylation of proteins may exert an effect on mitochondrial fission, Aβ production and synaptic damage, which may directly or indirectly contribute to Alzheimer’s disease pathology. In this paper, we review recent findings of an S-nitrosylated protein relationship to the pathogenesis of Alzheimer’s disease; discuss the influences of protein S-nitrosylation on Alzheimer’s disease progression; and consider indications for protein S-nitrosylation in the treatment of Alzheimer’s disease.Keywords
Alzheimer’s disease; Protein S-nitrosylation; Mitochondrial dysfunction; Synapse loss
Neuropathology of Alzheimer’s Disease
The glutamatergic and cholinergic systems are highly involved in memory and cognition. Dysregulation of these two systems is implicated in the progression of AD [15,16]. Glutamate is the main excitatory neurotransmitter in the central nervous system. Hyperactivation of N-methyl-D-aspartate (NMDA) glutamate receptor mediates excitotoxicity that contributes to AD pathology [15]. Non-competitive NMDA receptor antagonist memantine is currently used for the treatment of moderate-to-severe AD [17]. Degeneration of cholinergic neurons in basal forebrain has long been reported in AD patients [18], and cholinergic deficits lead to reduced synthesis of acetylcholine [18]. Therefore, a category of currently available drugs for mild to moderate stage of AD are used to enhance acetylcholine levels in the brain, including, for instance, donepezil, rivastigmine and galantamine [19].
Nitric oxide (NO) is produced from L-arginine catalyzed by nitric oxide synthases (NOS) including endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS) [16,20,21]. This process also requires cofactors/coenzymes such as nicotinamide adenine dinucleotide phosphate, flavine mononucleotide and flavin adenine dinucleotide [22,23]. NO participates in various cellular signaling pathways to regulate a spectrum of brain functions such as neuronal development, synaptic plasticity and apoptosis [24,,25]. The actions of NO are multifaceted and mainly classified into two categories: cyclic guanosine monophosphate (cGMP) dependent actions and cGMPindependent actions [26]. Studies have shown that NO at nano molar concentrations is sufficient enough to activate guanylate cyclase and trigger cGMP-dependent signals [26]. NO activated cGMP has been recognized to play critical roles in NO-mediated vasodilation [27]. In addition, NO/cGMP also contributes to the immune system, neurotransmission processes and regulation of cardiac contractility [28,30]. NO at higher than 50-100 μM can modify the thiol group of cysteine residues in proteins via covalently binding and induce protein cysteine S-nitrosylation [26,30,31]. Like other posttranslational modifications, S-nitrosylation is able to trigger conformational changes of various proteins. S-nitrosylatedthiols in cysteine residues can be reduced back to free thiols by glutathioneorthioredoxin[37]. For instance, S-nitrosothiol group can be denitrosylated by glutathione, forming a reduced protein thiol and S-nitrosoglutathione. S-nitrosoglutathione is rapidly and irreversibly metabolized by S-nitrosoglutathione reductase to glutathione S-hydroxysulfenamide. S-nitrosylatedthiols can also be denitrosylated by thioredoxin through its dithiol moiety to form thioredoxin disulfide and nitroxyl or NO, while thioredoxin disulfidecan be reduced back to thioredoxin by thioredoxin disulfide reductase [37].
Nitric Oxide and Protein S-nitrosylation
Cysteine residues in proteins are critical in regulating enzymes, transcription factors, protein structures and metal binding. Thiols of cysteine residues are very susceptible to be oxidatively modified by NO radicals. In AD, Aβ toxicity can cause the hyperactivity of NMDA receptors, resulting in increased Ca2+ influx that activates nNOS and increases NO production [a href="#ref38">38,39]. In addition, Aβ can increase NO production by activating iNOS in glial cells [28,35,40]. NMDA plays a critical role in the regulation of NO production in AD [41]. Increased NO in AD brains may target a wide variety of proteins. Indeed, many studies have presented compelling evidence implicating NO-inducedS-nitrosylation of proteins in the pathogenesis of AD [42-44].
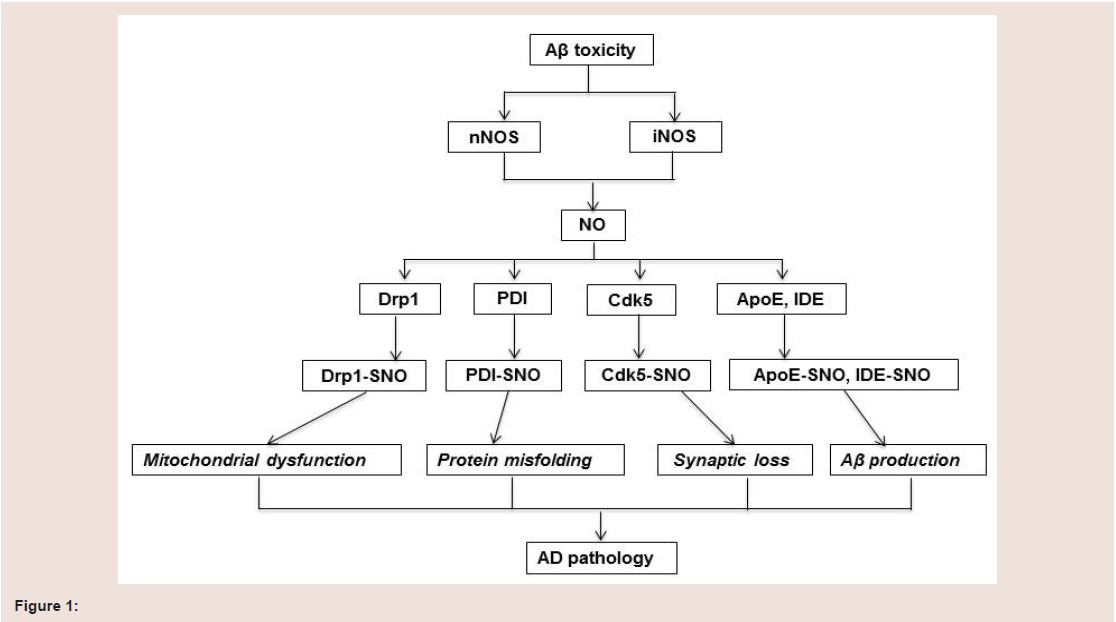
Many studies have shown that protein S-nitrosylation is increased in brain of AD patients. Although the exact mechanism of increased S-nitrosylation protein remains largely unknown, the vast majority of S-nitrosylated proteins are reported to be involved in mitochondrial dysfunction, protein misfolding, synaptic loss and Aβ production (figure 1).
Mitochondria are the main production sites for the cellular energy currency adenosine triphosphate. Mitochondrial functioning and energy metabolism are impaired early in the course of AD [45] but this mechanism is not fully understood. It has been reported that excessive S-nitrosylation of mitochondrial proteins may suppress protein functioning, thus compromising, at least partially, mitochondrial functioning, especially in neurodegenerative diseases such as AD where ROS/RNS are overwhelmed at a certain stage of the diseaseRecent studies show that S-nitrosylation of dynamin-related protein 1 (Drp1) is increased in AD. Drp1 is responsible for mitochondrial division and involved in regulation of mitochondrial fission. Mitochondrial functioning requires highly intact structure, in which an intricate balance of fusion and fission is a major determinant. Mitochondrial fusion promotes the mixing of organellar contents and unifies the mitochondrial compartment, both of which guarantee the exchange of mitochondrial DNA as well as various metabolites. Therefore, mitochondrial fusion ensures efficient production of adenosine triphosphate [46]. Fission, on the other hand, breaks down the unified mitochondrion into numerous small morphologically and functionally distinct organelles, leading to mitochondrial fragmentation, a signal of apoptosis [47]. Therefore, the imbalance between mitochondrial fusion and fission contributesto neurodegenerative diseases [48]. Drp1 as a dynamin-related guanosine triphosphatase mediates membrane fission through promoting dynamin dimerization and is a core component of the mitochondrial division machinery and responsible for excessive mitochondrial fission [49,50]. Cho et al. reported that S-nitrosylation of Drp-1 was increased both in mouse cerebrocortical cells treated with oligomeric Aβ25-35 and in post mortem brains of patients with sporadic AD [42]. Furthermore, Drp1 S-nitrosylation was also increased inhuman embryonic kidney (HEK) 293 cells transfected with nNOS, and this modification can be blocked by NOS inhibitor N-nitro-L-arginine [42]. This research group also found that Drp1 could not be S-nitrosylated when the Cys644 residue was mutated to Ala (C644A) [42], suggesting that Cys644 is the site for S-nitrosylation. Furthermore, NO donors could increase dynamin dimerization and guanosine triphosphatase activity, but not in Drp1 (C644A)mutated cells. These results suggest that S-nitrosylation of Cys644 residue in Drp1 inhibits Drp1 functioning. Aβ oligomers along withNO donors subsequently accelerated mitochondrial fragmentation and neuronal synaptic damage in HEK 293 cells [42]. These results suggest that S-nitrosylation of Drp1has a potential detrimental effect on mitochondrial dysfunction in AD.
The Role of S-nitrosylation in AD
S-nitrosylation on mitochondrial dysfunction
Relative levels of all proteins were determined by immunoblot analysis as previously described [35]. In short, proteins were resolved by SDS-PAGE with a total of 10 µg protein loaded into each well of the gel. Separated proteins were then transferred to polyvinylidene fluoride membranes (Bio-Rad). Membranes were probed with antiNWASP (1:1000; ab23394 Abcam), anti-WAVE1 (1:2500; ab50356 Abcam), anti-MBP (1:1000; ab53294 Abcam), and anti-MOG (1:10000; MAB5680 Millipore) primary polyclonal or monoclonal antibodies. Visualization and quantification of immunoblot bands was performed using the Gel Logic 2200 Pro (Carestream Molecular Imaging; Rochester, NY, USA). Samples were loaded in a randomized order with even numbers of PCP and control samples per time point per gel to minimize the effects of gel-to-gel variability on the results. A pooled sample combining aliquots from all 36 rats, was used as a positive control and was loaded onto each gel within the experiment to account for any gel-to-gel variability. Samples from each gel were then normalized to their respective pooled sample, and all immunoblot bands were normalized to a β-actin (1:5000; MAB1501 Millipore) same lane loading control. Mean β-actin expression levels did not differ between PCP and control groups (p>0.05). All experiments and quantifications were performed blind to treatment and age group.
Zahid et al. also reported that in post mortem hippocampus, substantia nigra and cortex of AD patients superoxide dismutase type 2 can be S-nitrosylated and that superoxide dismutase type 2S-nitrosylation is increased [13]. Superoxide dismutase type 2 is localized in mitochondria and is an antioxidant metalloenzyme that plays a critical role against oxidative and nitrosative stress in the mitochondrion [51,52]. These finding also supports that S-nitrosylation can contribute in part to mitochondrial dysfunction identified in AD.
S-nitrosylation on protein misfolding
Takashi et al. reported that PDI could be S-nitrosylated in HEK- 293 cells treated with NO donor S-nitrosocysteine (SNOC) or in the HEK-293 cells transfected with nNOS [56]. They further observed that S-nitrosylation of PDI was increased in post mortem brains of subjects with AD when compared to controls [56]. To determine whether S-nitrosylation affects PDI function, they monitored PDI chaperone and isomerase activities, which are two indexes for normal PDI functioning. Chaperone activity was assessed by measuring the aggregation of rhodanese induced by guanidinium. PDI significantly suppressed the aggregation of rhodanese, while S-nitrosyalted PDI also inhibited rhodanese aggregation, but with much lower efficiency [56]. Isomerase activity was then investigated by assessing the renaturation ability of PDI, in which a substrate of an inactivated form of RNase A containing scrambled disulfide bonds was used. The renaturation ability was two-fold in the wild type PDI over the S-nitrosylated PDI, indicating a reduced enzyme activity caused by S-nitrosylation [56]. Therefore, S-nitrosylation can inhibit the PDI enzyme activity and its chaperone activity. Given the major role that PDI plays in endoplasmic reticulum protein folding, it is likely that S-nitrosylation of PDI may impair the protein folding process and exacerbate endoplasmic reticulum stress, ultimately leading to cell death in AD.
Neuronal dysfunction and degeneration in AD patients is predominantly in the hippocampus and cerebral cortex [6]. Cyclindependent kinase 5 (Cdk5) plays an important role in various neuronal functions including axon guidance, neuronal migration, neuronal survival and the regulation of synaptic spine density [57-60]. Dysregulation of Cdk5 activity contributes significantly to the pathogenesis of neurodegenerative disorders. Hyperactivity of Cdk5 triggered by oxidative stress, Aβ and calcium overload contributes toneuronal death in AD [43].
S-nitrosylation on synaptic loss
It has been found that NO donor SNOC increased S-nitrosylation of Cdk5 and Cdk5 enzyme activity in cultured cells [43]. S-nitrosylation sites were further identified at Cys83 and Cys157 residues in Cdk5 [43]. SNOC only increased the protein activity in wild type-Cdk5 cells but not in mutant-Cdk5 cells [43]. This result indicates that S-nitrosylation of Cdk5 increases Cdk5 enzyme activity. Cdk5 is an important modulator of NMDA receptor signaling and neuronal spine loss [43].It has been found that NMDA can increase Cdk5 S-nitrosylation, which can be abrogated by NOS inhibitor N-nitroarginine, indicatingCdk5 S-nitrosylation is mediated by NMDA receptor. NMDA also more significantly increased spine loss in the wide type-Cdk5/p35 cellsthan in mutant-Cdk5/p35 cells, suggesting that Cdk5 S-nitrosylation contributes to NMDA-induced dendritic spine loss [43]. Exposure of Aβ1-42 oligomers induces S-nitrosylation of Cdk5 in cultured ratcortical neurons. This treatment also induces loss of dendriticspines in neurons transfected with Cdk5/p35, but not in mutant-Cdk5/p35. Aβ-induced Cdk S-nitrosylation and spine loss can be completely abolished by adding NOS inhibitor N-nitroarginine, suggesting that Cdk5 S-nitrosylation in AD contributes to the spine loss through NMDA receptors [43]. Recently it was found that S-nitrosylation of Cdk5 can act as an endogenous nitrosylase for Drp1 and induce Drp 1 S-nitrosylation by transferring NO group from S-nitrosylated Cdk5to Drp1, contributing to mitochondrial dysfunction and synapse loss [43]. All of these findings suggest that increased S-nitrosylation of Cdk5 changes its enzyme activity and affects the functioning of otherproteins that may contribute to Aβ/NMDA receptor-mediated spine loss and the pathogenesis of AD [43]. In addition both alpha and betatubulins were identified as targets for S-nitrosylation in human AD postmortem brains [13] that may also contribute to synaptic loss in AD. The functional changes from this S-nitrosylation are not entirely clear. However, since alpha and beta tubulins play an important role in microtubule structure or cellular architecture, S-nitrosylation of these proteins may be a potential target therapy for the future study.
Beta-site APP cleaving enzyme 1 (BACE1), insulin-degrading enzyme (IDE) and apolipoprotein E (ApoE) can regulate Aβ metabolism and aggregation. Studies have shown that these three proteins can all be S-nitrosylated and that S-nitrosylation of BACE1, IDE and ApoE are seen to be increased in both cell models and AD post mortem brains [26,61,62], suggesting that protein S-nitrosylation may affect Aβ production and deposition.
S-nitrosylation on Aβ production
BACE1 together with γ-secretase cleaves transmembrane APP to form b-amyloid. Since BACE1 is essential for Ab production, the expression level and enzymatic activity of BACE1 can change the Aβ deposit. Young et al found that NO donor SNOC increased S-nitrosylation of BACE1and decreased BACE1 activity in primary cultured cortical neurons [26]. BACE1 S-nitrosylation in AD brains was higher in the mild cognitive impairment stage than in the late stage, which inversely correlates with BACE1 protein levels [26]. These findings together demonstrate a suppressive effect on Aβ load from S-nitrosylation of BACE1.
IDE is a conserved zinc metalloprotease that is responsible for the clearance of various hormones and peptides including insulin and Aβ [63]. It has been evidenced that the NO donor S-nitroso- N-acetylpenicillamine could increase S-nitrosylation of purified rat IDE enzyme and also increase IDE S-nitrosylation in HEK 293T cells transfected with human IDE [61]. Cys110, Cys178, Cys789, Cys819 and Cys966 residues of IDE were identified to be S-nitrosylated. S-nitrosylation of Cys110 and Cys819 leads to complete inactivation of IDE. S-nitrosylation of both Cys789 and Cys966 together also causes conformational change and aggregation of IDE, resulting in the inhibition of IDE activity [63]. It was found that various NO donors could significantly reduce IDE enzyme activity, and insulin and Aβ degradation [61]. This result suggests a potential role of S-nitrosylation on IDE function and Aβ production. Since IDE is a very important modulator for Aβ production, inhibition of IDE activity by NO in the brain may increase the accumulation of Aβ, leading to neuronal death.
ApoE represents a family of proteins including ApoE2 ApoE3 and ApoE4, which are expressed in the brain [64]. ApoE is essential for the normal catabolism of triglyceride-rich lipoprotein metabolites. ApoE isoforms enhance the break-down of Aβ, while ApoE4 is not as effective as the others at promoting these reactions. Therefore gene variation of ApoE4 normally results in increased vulnerability to AD, making it one of the important genetic risk factors for AD pathology [65]. Alexander et al. reported that S-nitrosylation of ApoE2 and ApoE3, but not ApoE4 is increased in the HEK-293 cell line transiently transfected with nNOS, suggesting that ApoE2 and ApoE3 can be S-nitrosylated [62]. Specifically, Cys112 and Cys158 in ApoE2 and Cys112 in ApoE3 were identified as S-nitrosylation sites. S-nitrosylation of ApoE2 and ApoE3 inhibits the binding of ApoE to low density lipoprotein receptors[62]. Since the activities of the ApoE isoforms are essential for the break-down of Aβ, the inhibitory effect caused by S-nitrosylation on ApoE2 and ApoE3 may potentially increase Ab deposit, contributing to AD pathology.
Aβ plaque, as one of the most important pathological hallmarks in AD, contributes to NMDA receptor hyperactivity, synapse loss and neuronal cell death. NO may regulate Aβ production by S-nitrosylating proteins. In these findings, BACE1 S-nitrosylation potentially suppresses Aβ production, showing a ‘beneficial’ effect, while IDE S-nitrosylation and ApoE S-nitrosylation potentially increase Aβ production, showing a ‘detrimental’ effect. These results indicate a potential pharmaceutical way to reduce Aβ production either by S-nitrosylating beneficial proteins or denitrosylating detrimental proteins.
Many S-nitrosylated proteins have recently been identified in AD post mortem brains or AD animal models by high throughput proteomic studies techniques with mainly S-nitrosylated protein enrichment and mass spectrometry [50]. WEB-based Gene SeT AnaLysis Toolkit (WEBGESTALT) and UniProt show that the vast majority of S-nitrosylated proteins are related to signaling pathways, including apoptosis, energy metabolism and redox regulation [13]. In addition, 138 synaptic proteins have been found to be S-nitrosylated in a transgenic animal model, APPV717I [14]. The APPV717I animal model is an AD mouse model over expressing human APP with the London mutation driven by the Thy1 promoter. This finding indicates that S-nitrosylation may have a potential role in mediating neurotransmission
Indications for the S-nitrosylation process in the pharmacological treatment of AD
Oxidative stress is one of the main characteristics in Alzheimer’s disease. NO can induce protein S-nitrosylation, Protein S-nitrosylation is known to be involved in mitochondrial dysfunction, protein misfolding, synapse loss and Aβ production and therefore may have either a direct or an indirect effect on AD pathology. These findings suggest that the process of S-nitrosylation could be a good target for the pharmacological treatment of AD. These unique characteristics ofS-nitrosylation are a significant potentiality for developing new drugs for AD patients.
Antioxidants that can scavenge the excessive NO within the brain may have potential for AD treatment involving S-nitrosylation. Polyphenolic compounds extracted from green tea, magnolia, blueberries and grapes scavenge ROS/RNS including NO and exhibit neuroprotective effects. Studies have shown that these compounds produce beneficial effect on both in vivo and in vitro in animal models for AD [66-70]. Since inhibition of NOS can reduce NO production, NOS inhibitors may also be beneficial for AD treatment. NOS inhibitors 1400W (N-(3-(aminomethyl)benzyl) acetamidine), L-NIL (N-iminoethyl-L-lysine) and L-NAME (N-nitro-L-arginine methylester) were found not only to reduce NO release but also to inhibit Aβ and glutamate-induced neuronal cell death [71-73]. Other compounds have also demonstrated a beneficial effect on AD pathology by inhibiting NOS activity. Rutin is a flavonoid that can dose-dependently decrease Aβ42-induced cytotoxicity in human SHSY5Y neuroblastoma cells. One of its main effects is to decrease NO production by reducing iNOS activity [73]. Ferulic acid ethyl ester was reported to protect neurons against Aβ1-42-induced toxicity in primary neuronal cell culture in part by down-regulation of iNOS [74]. Xanthorrhizol reduced ROS generation and glutamate-induced neurotoxicity in the murine hippocampal HT22 cell line, partially by reducing the expression of iNOS, which consequently resulted in the reduction of NO [71].
NMDA receptors are now considered to be a target for AD treatment as it plays vital role in neuronal excitotoxicity. NMDA receptor activation can increase NO production and that increased NO can then nitrosylate NMDA receptor, resulting in negative feedback regulation of NMDA receptor activity. Therefore, alteration of NMDA receptor status may also have a potential for AD treatment. Nitroglycerin, which donates NO, has been shown to limit excessive NMDA receptor activity in rat AD models [73,75]. Nitromemantines are combinatorial drugs of memantine and NO donors. Theoretically memantine works as a homing molecule to allow NO donor to combine with S-nitrosylation sites of the NMDA receptor. Nitromemantines have been proven to be highly neuroprotective in cultured rat primary cortical neurons treated with Aβ [76].
Protein S-nitrosylation contributes significantly to the pathological development of AD. Protein S-nitrosylation occurs in response to excessive NO production, which may regulate mitochondrial dysfunction, protein misfolding, synaptic loss and Aβ production. The global S-nitrosylation of proteins detected in AD indicates a potential pharmaceutical way forward to development of future AD therapies. However, our knowledge of the molecular mechanisms of S-nitrosylation in AD is by no means complete. In future studies, more work will be needed to elucidate the precise mechanisms of S-nitrosylated proteins in AD pathology. In all, it is an interesting and promising topic for future AD therapies.
Conclusions
References
- Kenche VB, Barnham KJ (2011) Alzheimer’s disease & metals: therapeutic opportunities. Br J Pharmacol 163: 211-219.
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2004) State-specific projections through 2025 of Alzheimer disease prevalence. Neurology 62: 1645.
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, et al. (1991) Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353: 844-846.
- LaFerla FM, Green KN, Oddo S (2007) Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci 8: 499-509.
- Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298: 789-791.
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, et al. (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572-580.
- Kumar S, Walter J (2011) Phosphorylation of amyloid beta (Aβ) peptides-a trigger for formation of toxic aggregates in Alzheimer's disease. Aging 3: 803-812.
- Gozes I (2010) Tau pathology and future therapeutics. Curr Alzheimer Res 7: 685-696.
- Chun W, Johnson GV (2007) The role of tau phosphorylation and cleavage in neuronal cell death. Front Biosci 12: 733-756.
- Crews L, Masliah E (2010) Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet 19: R12-20.
- Tiraboschi P, Sabbagh MN, Hansen LA, Salmon DP, Merdes A, et al. (2004) Alzheimer disease without neocortical neurofibrillary tangles: a second look. Neurology 62: 1141-1147.
- Sayre LM, Perry G, Smith MA (2008) Oxidative stress and neurotoxicity. Chem Res Toxicol 21: 172-188.
- Zahid S, Khan R, Oellerich M, Ahmed N, Asif AR (2013) Differential S-nitrosylation of proteins in Alzheimer's disease. Neuroscience 256: 126-136.
- Zaręba-Kozioł M, Szwajda A, Dadlez M, Wysłouch-Cieszyńska A, Lalowski M (2014) Global analysis of S-nitrosylation sites in the wild type (APP) transgenic mouse brain-clues for synapticpathology. Mol Cell Proteomics 13: 2288-2305.
- Olney JW, Wozniak DF, Farber NB (1997) Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch Neurol 54: 1234-1240.
- Bredt DS, Snyder SH (1990) Isolation of nitric oxide synthetase, a calmodulin- requiring enzyme. Proc Natl Acad Sci U S A 87: 682-685.
- Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, et al. (2004) Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 291: 317-324.
- Contestabile A (2011) The history of the cholinergic hypothesis. Behav Brain Res 221: 334-340.
- Rossignol DA, Frye RE (2014) The use of medications approved for Alzheimer's disease in autism spectrum disorder: a systematic review. Front Pediatr 2: 87.
- Popp R, Fleming I, Busse R (1998) Pulsatile stretch in coronary arteries elicits release of endothelium-derived hyperpolarizing factor: a modulator of arterial compliance. Circ Res 82: 696-703.
- Dawson VL, Dawson TM (1998) Nitric oxide in neurodegeneration. Prog Brain Res 118: 215-229.
- Knowles RG, Moncada S (1994) Nitric oxide synthases in mammals. Biochem J 298: 249-258.
- Stuehr DJ (1999) Mammalian nitric oxide synthases. Biochim BiophysActa 1411: 217-230.
- Dreyer J, Schleicher M, Tappe A, Schilling K, Kuner T, et al. (2004) Nitric oxide synthase (NOS)-interacting protein interacts with neuronal NOS and regulates its distribution and activity. J Neurosci 24: 10454-10465.
- Fukui H, Moraes CT (2008) The mitochondrial impairment, oxidative stress and neurodegeneration connection: reality or just an attractive hypothesis? Trends Neurosci 31: 251-256.
- Kwak YD, Wang R, Li JJ, Zhang YW, Xu H, et al. (2011) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener 6: 17.
- Tanaka Y, Tang G, Takizawa K, Otsuka K, Eghbali M, et al. (2006) Kv channels contribute to nitric oxide- and atrial natriuretic peptide-induced relaxation of a rat conduit artery. J Pharmacol Exp Ther 317: 341-354.
- Hopper RA, Garthwaite J (2006) Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci 26: 11513-11521.
- Taqatqeh F, Mergia E, Neitz A, Eysel UT, Koesling D, et al. (2009) More than a retrograde messenger: nitric oxide needs two cGMP pathways to induce hippocampal long-term potentiation. J Neurosci 29: 9344-9350.
- Green SJ (1995) Nitric oxide in mucosal immunity. Nat Med 1: 515-517.
- Lipton SA, Singel DJ, Stamler JS (1994) Nitric oxide in the central nervous system. Prog Brain Res 103: 359-364.
- Foster MW, Hess DT, Stamler JS (2009) Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med 15: 391-404
- Drew B, Leeuwenburgh C (2002) Aging and the role of reactive nitrogen species. Ann N Y Acad Sci 959: 66-81.
- Wink DA, Miranda KM, Espey MG (2000) Effects of oxidative and nitrosative stress in cytotoxicity. Semin Perinatol 24: 20-23.
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, et al. (1992) Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8: 1087-1099.
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, et al. (2013) Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 78: 596-614.
- Benhar M, Forrester MT, Stamler JS (2009) Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 10: 721-732.
- Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, et al. (1991) Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351: 714-718.
- Garthwaite J, Charles SL, Chess-Williams R (1988) Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336: 385-388.
- Zhao QF, Yu JT, Tan L (2015) S-Nitrosylation in Alzheimer's disease. Mol Neurobiol 51: 268-280.
- Gu Z, Nakamura T, Lipton SA (2010) Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction inneurodegenerative diseases. Mol Neurobiol 41: 55-72.
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, et al. (2009) S-Nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324: 102-105.
- Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, et al. (2011) S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc Natl Acad Sci U S A 108: 14330-14335.
- Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, et al. (2011) S-Nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron 71: 131-141.
- Westermann B (2009) Nitric oxide links mitochondrial fission to Alzheimer's disease. Sci Signal 2: pe29.
- Westermann B (2002) Merging mitochondria matters: Cellular role and molecular machinery of mitochondrial fusion. EMBO Rep 3: 527-531.
- Youle RJ, Karbowski M (2005) Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol 6: 657-663.
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505-518.
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, et al. (2006) Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25: 3900-3911.
- Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, et al. (2008) Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: 19318-19323.
- Murakami K, Shimizu T (2012) Cytoplasmic superoxide radical: a possible contributing factor to intracellular Abetaoligomerization in Alzheimer disease. Commun Integr Biol 5: 255-258.
- Fukai T, Ushio-Fukai M (2011) Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 15: 1583-1606.
- Lyles MM, Gilbert HF (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: pre-steady-state kinetics and the utilization of the oxidizing equivalents of the isomerase. Biochemistry 30: 619-625.
- Lyles MM, Gilbert HF (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30: 613-619.
- Kaufman RJ (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13: 1211-1233.
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, et al. (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513-517.
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, et al. (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A 93: 11173-11178.
- Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH (2003) Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114: 469-482.
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, et al. (2006) Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442: 814-817.
- Angelo M, Plattner F, Giese KP (2006) Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem 99: 353-370.
- Cordes CM, Bennett RG, Siford GL, Hamel FG (2009) Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem Pharmacol 77: 1064-1073. .
- Abrams AJ, Farooq A, Wang G (2011) S-nitrosylation of ApoE in Alzheimer's disease. Biochemistry 50: 3405-3407.
- Malito E, Ralat LA, Manolopoulou M, Tsay JL, Wadlington NL, et al. (2008) Molecular bases forthe recognition of short peptide substrates and cysteine-directed modifications of humaninsulin-degrading enzyme. Biochemistry 47: 12822-12834.
- Ghebranious N, Ivacic L, Mallum J, Dokken C (2005) Detection of ApoE E2, E3 and E4 alleles using MALDI-TOF mass spectrometry and the homogeneous mass-extend technology. Nucleic Acids Res 33: e149.
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, et al. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58: 681-693.
- Choi DY, Lee YJ, Hong JT, Lee HJ (2012) Antioxidant properties of natural polyphenols and their therapeutic potentials for Alzheimer's disease. Brain Res Bull 87: 144-153.
- Fu X, Zhang J, Guo L, Xu Y, Sun L, et al. (2014) Protective role of luteolin against cognitive dysfunction induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav 126: 122-130.
- Brondino N, Re S, Boldrini A, Cuccomarino A, Lanati N, et al. (2014) Curcumin as a therapeutic agent in dementia: a mini systematic review of human studies. ScientificWorldJournal 2014: 174282.
- Zhang SQ, Sawmiller D, Li S, Rezai-Zadeh K, Hou H, et al. (2013) Octylgallate markedly promotes anti-amyloidogenic processing of APP through estrogen receptor-mediated ADAM10 activation. PLoS One 8: e71913.
- Lee YW, Kim DH, Jeon SJ, Park SJ, Kim JM, et al. (2013) Neuroprotective effects of salvianolic acid B on an Aβ25-35 peptide-induced mouse model of Alzheimer's disease. Eur J Pharmacol 704: 70-77.
- Perrella J, Bhavnani BR (2005) Protection of cortical cells by equine estrogens against glutamate-induced excitotoxicity is mediated through a calcium independent mechanism. BMC Neurosci 6: 34.
- Greco R, Amantea D, Blandini F, Nappi G, Bagetta G, et al. (2007) Neuroprotective effect of nitroglycerin in a rodent model of ischemic stroke: evaluation of Bcl-2 expression. Int Rev Neurobiol 82: 423-435.
- Javed H, Khan MM, Ahmad A, Vaibhav K, Ahmad ME, et al. (2012) Rutin prevents cognitive impairments by ameliorating oxidative stress and neuroinflammation in rat model of sporadic dementia of Alzheimer type. Neuroscience 210: 340-352.
- Perluigi M, Joshi G, Sultana R, Calabrese V, De Marco C, et al. (2006) In vivo protective effects of ferulic acid ethyl ester against amyloid-beta peptide 1-42-induced oxidative stress. J Neurosci Res 84: 418-426.
- Vámos E, Párdutz A, Varga H, Bohár Z, Tajti J, et al. (2009) l-kynurenine combined with probenecid and the novel synthetic kynurenic acid derivative attenuate nitroglycerin-induced nNOS in the rat caudal trigeminal nucleus. Neuropharmacology 57: 425-429.
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, et al. (2013) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A 110: E2518-2527.