
Journal of Pharmaceutics & Pharmacology
Download PDF
 Figure 1: Chemical structure of GLA.
Figure 1: Chemical structure of GLA.
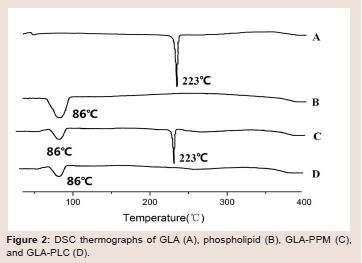 Figure 2: DSC thermographs of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
Figure 2: DSC thermographs of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
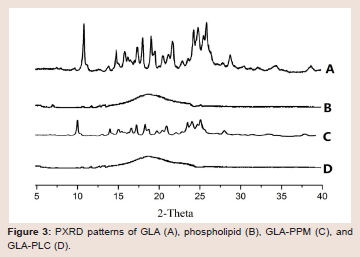 Figure 3: PXRD patterns of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
Figure 3: PXRD patterns of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
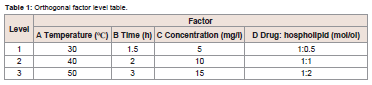 Table 1: Orthogonal factor level table.
Table 1: Orthogonal factor level table.
 Figure 4: SEM photomicrographs of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
Figure 4: SEM photomicrographs of GLA (A), phospholipid (B), GLA-PPM (C), and GLA-PLC (D).
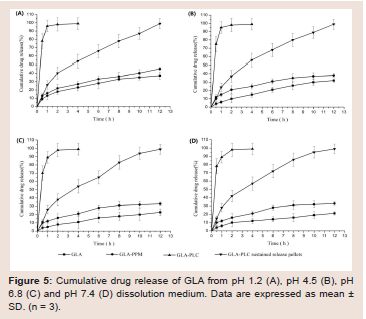 Figure 5: Cumulative drug release of GLA from pH 1.2 (A), pH 4.5 (B), pH 6.8 (C) and pH 7.4 (D) dissolution medium. Data are expressed as mean ± SD. (n = 3).
Figure 5: Cumulative drug release of GLA from pH 1.2 (A), pH 4.5 (B), pH 6.8 (C) and pH 7.4 (D) dissolution medium. Data are expressed as mean ± SD. (n = 3).
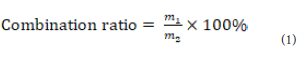
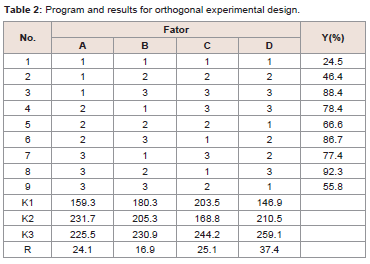 Table 2: Program and results for orthogonal experimental design.
Table 2: Program and results for orthogonal experimental design.
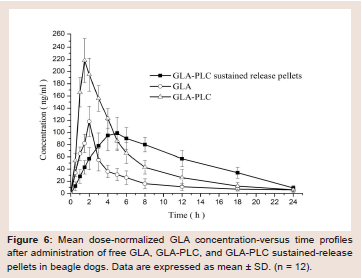 Figure 6: Mean dose-normalized GLA concentration-versus time profiles
after administration of free GLA, GLA-PLC, and GLA-PLC sustained-release
pellets in beagle dogs. Data are expressed as mean ± SD. (n = 12).
Figure 6: Mean dose-normalized GLA concentration-versus time profiles
after administration of free GLA, GLA-PLC, and GLA-PLC sustained-release
pellets in beagle dogs. Data are expressed as mean ± SD. (n = 12).
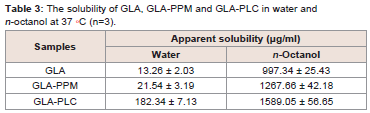 Table 3: The solubility of GLA, GLA-PPM and GLA-PLC in water and
n-octanol at 37 ◦C (n=3).
Table 3: The solubility of GLA, GLA-PPM and GLA-PLC in water and
n-octanol at 37 ◦C (n=3).
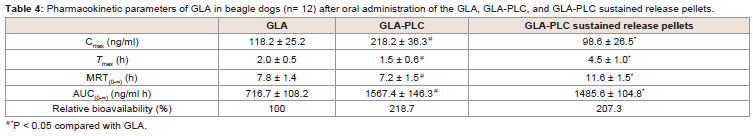 Table 4: Pharmacokinetic parameters of GLA in beagle dogs (n= 12) after oral administration of the GLA, GLA-PLC, and GLA-PLC sustained release pellets.
Table 4: Pharmacokinetic parameters of GLA in beagle dogs (n= 12) after oral administration of the GLA, GLA-PLC, and GLA-PLC sustained release pellets.
Research Article
Preparation and Evaluation of Glaucocalyxin A Sustained- Release Pellets Based on Phospholipid Complex System with Enhanced Bioavailability
Miao YF1* and Sun JQ2
1College of chemistry and chemical engineering, Taishan University, China
2Office of Research Affairs, Taishan University, China
*Address for Correspondence: Miao YF, College of chemistry and chemical engineering, Taishan University, Tai’an, China; E-mail: lanjin0309@163.com
Submission: 20 June 2020;
Accepted: 05 August 2020;
Published: 07 August 2020
Copyright: © 2020 Miao YF, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abstract
Objective:
Glaucocalyxin A (GLA) suffers from low oral bioavailability
and rapid in vivo metabolism. Therefore, the purpose of this study
was to develop a new formulation to enhance the oral bioavailability
simultaneously sustained release of GLA.
Material and methods:
GLA-phospholipid complex was firstly formulated
by solvent-evaporation method to improve the solubility of GLA.
Differential scanning calorimetry, powder X-ray diffraction, scanning
electron microscopy, and solubility study were used to characterize the
GLA-phospholipid complex. And then, the optimized GLA-phospholipid
complex was selected to prepare GLA-phospholipid complex
sustained release pellets by extrusion-spheronization and fluidized bed
coating technology. The prepared pellets were studied by in vitro drug
release study and administered to beagle dogs to evaluate the oral
bioavailability of GLA-phospholipid complex and GLA-phospholipid
complex sustained release pellets.
Results:
The results illustrated that GLA in GLA-phospholipid complex was either molecularly dispersed or in an amorphous form with 13.8-fold water solubility than the free GLA. The pharmacokinetic studies in beagle dogs demonstrated that GLA-phospholipid complex and GLAphospholipid complex sustained-release pellets showed 2.19-fold and 2.07-fold improvement than the free GLA by oral dosage, respectively.
Further, steady plasma concentration, and prolonger Tmax were simultaneously obtained from GLA-phospholipid complex sustained release pellet.
Conclusion:
These outcomes suggested that the combination of phospholipid complex and sustained release pellets could enhance the oral bioavailability and prolong the action time in vivo which provided a promising delivery system for GLA.
Keywords
Glaucocalyxin A; Sustained release pellet; Phospholipid complex; Bioavailability; Solubility
Abbreviations
GLA: Glaucocalyxin A; GLA-PLC 1: GLA-Phospholipid Complex; DSC: Differential Scanning Calorimetry; PXRD: PowderX-ray Diffraction; SEM: Scanning Electron Microscopy; GLA-PPM: GLA-Phospholipid Physical Mixture
Introduction
In the last two decades, about 40% of discovered active pharmaceutical ingredients either from the synthetic or natural origin suffers from low oral bioavailability [1-3]. This might be attributed to the low water solubility and/or permeability of drug across the biological membrane [4,5]. Glaucocalyxin A (GLA) is a biologically active ent-kaurane diterpenoid isolated from inulajaponica var, a traditional Chinese medicinal herb that grows in the northeastern of China [6,7]. The structure of GLA is shown in with molecular formula of 20H28O4 (Figure 1). GLA has been widely used in China folk medicine as cytotoxicity and antitumor, anti-bacterial, anti-inflammatory, stomachic and anthelmintic agent for more than 30 years [8].
However, there are two main disadvantages of GLA for its practical
use as a therapeutic agent, one is the poor water solubility caused
low oral bioavailability, the other is the rapid in vivo metabolism
which need the patient to take the medicine more than three times
a day [9,10]. To overcome the drawbacks, several strategies had
been proposed to increase the water solubility of GLA including
nanosuspensions [11], nanoparticle [12], inclusion complex [13], and self-nanoemulsion [14] etc.
Those researches demonstrated that when the GLA formulation
with a better solubility, GLA showed stronger antitumor activity
compared with the free drug [11,12]. Nevertheless, there was few
research focus on extending the action time of medicament within
human body along with improving the oral bioavailability of GLA. As
a result, there is an urgent demand to develop a new formulation with
prolonged actions and enhanced oral bioavailability of GLA.
In recent years, the technique of lipid-based drug delivery system, such as liposomes [15,16], solid lipid nanoparticles [17,18],
phytosomes etc. [19]. Have acquired more attention as they can
resolve major drawbacks related to drug delivery. However, there are
some shortcomings, like drug leakage, low drug loading, and poor
stability that remain to be a major concern for liposome. Similarly,
low inherent incorporation rate, high tendency for drug expulsion,
and unpredictable gelation tendency of solid lipid nanoparticles
restrict their overall utilization [20,21]. Besides that, the related
manufacturing process is sophisticated and difficult to scale up.
Among these, the drug-phospholipid complex has become one of
the most successful strategies for improving the oral bioavailability
of a number of poorly water soluble drugs by enhancing solubility,
permeability, and by improving metabolic stability in gastrointestinal
tract [22-24]. Being comprised with such property, phospholipid
complex is a suitable delivery vehicle for drugs which have limited
solubility (BCS class II) limited permeability (BCS class III), and both
(BCS class IV). Meanwhile, it is relatively cheap, easy to formulate,
and easy to scale up compared with other lipid-based drug delivery
systems [25].
Spherical free-flowing pellets, as multiple unit sustained release
drug reservoirs of narrow size distribution are popular in commercial
products. The pellets offer many clinical advantages compared with
single unit dosage forms, such as less variability release profiles [26],
stable plasma concentrations [27], reduced intra-and inter-subject
variability on drug plasma [28], decreased local irritations, less dose
dumping risk and great reproducibility [29].
Therefore, in this study, in order to enhance the oral bioavailability
and prolong the action time of GLA in vivo, we developed a novel
GLA-Phospholipid Complex (GLA-PLC) sustained release pellets
formulation to take advantage of the solubility and absorption
enhancing effect of phospholipid complex and release rate-controlling
capacity of the sustained release pellets.
Materials and Methods
Materials:
Glaucocalyxin A was obtained from College of Pharmaceutical
Science, Soochow University (Suzhou, China). Lipoid E80 (trade
name of phospholipid)was purchased from Shanghai Dongshang
BiologyTechnique Ltd. (Shanghai, China). Lactosewas purchased
from Meggle. (Germany). Kollicoat SR 30 D was purchased from
BASF (Germany). Unless otherwise stated, all other materials were
of analytical grade.
Preparation of GLA-PLC:
Solvent evaporation method was applied to prepare GLA-PLC.
In short, GLA-PLC was prepared with drug and phospholipid
in 1:0.5, 1:1, 1:2, or 1:3 molar ratios. Accurate quantity of GLA
and phospholipid were taken in a 250 mL round bottom flask and
dissolved in anhydrous ethanol to obtain uniform solution with the
drug concentration of 2 mg/ml, 5 mg/ml, 10 mg/ml, 15 mg/ml, or 20
mg/ml. The prepared mixture solution was refluxed at 25 0C, 30 0C,
40 0C, 50 0C or 60 0C for 0.5 h, 1 h, 1.5 h, 2 h, 3 h or 4 hunder constant
stirring at 100 rpm to form GLA-PLC. Subsequently, anhydrous
ethanol was evaporated by rotary evaporation under reduced pressure
to give solid product.
Based on the single factor experiment, GLA-PLC preparation conditions were optimized by L9 (34) orthogonal experiment, which
can be found in Table 1. The dried residues were placed in a desiccator
in room temperature until used.
The same ratio of GLA and phospholipid was simply mixed to
form GLA-phospholipid physical mixture (GLA-PPM). The resulting
samples was stored in a desiccator in room temperature until used.
The combination ratio of GLA-PLC:
The combination ratio of GLA to phospholipid was measured
under the difference of solubility between GLA and GLA-PLC [30].
GLA-PLC could be dissolved in n-hexane, but free GLA could not be
dissolved in n-hexane. The combination ratio was calculated on the
following basic equation (Eq. (1):
Where, m1 is the combination amount of GLA in the GLA-PLC,
and m2 is the total amount of GLA in the GLA-PLC.
Characterization of GLA-PLC:
Determination of GLA content in phospholipid complex: The
content of GLA in the complex was determined by reversed-phase
HPLC method (Agilent 1260, Agilent Technologies, Inc., USA)
using a Phenomenex Luna C18 column (4.6 mm × 250 mm, 5μm)
and a 1.0 ml/min flow rate, a 20 μl injection volume, a 40 0C column
temperature, 231 nm UV detection. The mobile phase was comprised
of acetonitrile and water in a volume ratio of 35:65.
Solubility studies:
Solubility determination of GLA, GLAPPM,
and GLA-PLC were carried out by adding excess of the above
samples to 10 ml of distilled water and n-octanol in sealed glass vials,
respectively. Each experiment was performed in triplicate. After 12
h of shaking in the shaker at 100 rpm under 37 0C, the solution was
centrifuged at 10,000 rpm for 5 min and the obtained supernatants
were filtered (0.45 μm, Jinlong, Tianjin, China). The concentration
of GLA in the resulting solution was diluted with mobile phase and
analyzed by HPLC as described in “2.4.1 Determination of GLA content in phospholipid complex”.
Differential scanning calorimetry (DSC):
Thermal characteristic
of GLA, phospholipid, GLA-PPM, and GLA-PLC were determined
using an ATA449-C instrument (NETZSCH, Selb, Germany) under
a 10 ml/min stream of nitrogen gas flow. About five milligrams of the
samples were heated from 40 0C to 400 0C at the heating rate of 10 0C
/min in an open aluminum pan.
Powder X-ray diffraction (PXRD):
The PXRD patterns of GLA,
phospholipid, GLA-PPM, and GLA-PLC were obtained on a D/MAX
2500-PC X-ray powder diffraction meter (Rigaku Denki, Tokyo,
Japan), with a voltage of 50 kV and a current of 40 mA. Data was
scanned from 5° to 40° (2θ) with a step size of 0.02°.
Scanning electron microscopy (SEM):
The surface morphology
of GLA, phospholipid, GLA-PPM, and GLA-PLC were examined
using anS-4100 scanning electron microscope (Hitachi, Tokyo,
Japan). Samples were sputter coated with gold-palladium and
observed at different magnifications.
Preparation of GLA-PLC sustained release pellets:
GLA-PLC sustained release pellets were conducted by extrusionspheronization
and fluidized bed coating technology by the following steps: first, GLA-PLC was mixed with lactose in 1:0.5 weight ratio to
get free flowing powder and then sieved by a 60 mesh. Second, the
prepared powder was moistened by purified water in order to get
soft material. This soft material was extruded by extruder (Guanlian
Pharmaceutical Equipment Co., Ltd., Shanghai, China) with dies of
1.0 mm diameter and extrusion speed of 30 rpm. The extrudates were
then placed in a spheronizer fitted with a cross-hatched plate rotated
for 15 min at a spheronization speed of 1000 rpm. The spheronized
pellets were sieved after had been dried in fluidized bed processor
(Niro-Aeromatic, Switzerland) at 50 0C for 25 min.
Finally, the pellets were coated through fluidized bed coating
technology as described by PateL SA et al. with some modifications
[31]. The coating polymer suspension was prepared by adding16 g
triethyl citrate (as plasticizer) and 530 g Kollicoat SR 30 D to 430 g
purified water with stirring for 40 min. 55 g talc was added to the
above suspension as an anti-tacking agent. The coating suspension
was stirred during the coating process. The coating was done at
10% weight gain using a laboratory-scale fluid bed coater (Hanse,
Changzhou, China).Experimental parameters were pre-warming of
core pellets at 40 0C for 10 min; atomizing air pressure 1.2 bars; spray
nozzle diameter 1.2 mm; air flow rate 90 m3.h-1; inlet air temperature
50-55 0C; product temperature 35-40 0C; spray rate 0.5 ml.min-1; post
drying at 55 0C for 15min.
<em>In vitro</em> drug release:
The drug release profiles of GLA, GLA-PPM, GLA-PLC, and
GLA-PLC sustained-release pellets (all those formulations contained
20 mg GLA were filled into hard gelatin capsules) were studied using
Chinese Pharmacopoeia type Ⅲ Apparatus (Tianjin Tianda Tianfa
Technology Co., Ltd, Tianjin, China), paddle method with big vessel,
at the paddle rotation speed of 50 rpm in 900 ml dissolution medium,
at 37 ± 0.5℃. At the predetermined times, 1 ml aliquot was withdrawn,
filtered (0.45 μm, Jinlong, Tianjin, China), and analyzed by reversedphase
HPLC method as described in “2.4.1 Determination of GLA
content in phospholipid complex”. 1.0 ml of fresh dissolution media
pre-warmed to 37 0C 0.5 0C was replaced into the dissolution medium
after each sampling. All the experiments were performed in triplicate.
Pharmacokinetic studies in dogs:
The in vivo pharmacokinetic study described here was reviewed
and approved by the Nanjing Medical University Institutional
Animal Care and Use Committee (No. 2019-0306) and adhered to the
“Principles of Laboratory Animal Care”. Tofacilitate administration,
GLA, GLA-PLC, and GLA-PLC sustained release pellets were filled
into hard gelatin capsules, each dosage contained 20 mg GLA.
An open label, randomized, three-period crossover experiment
design was adopted to evaluate the pharmacokinetics of the prepared
capsules. Twelve male beagle dogs weighting 10 ± 1 kg were divided
into three group and tested under fasted state (with free access to water). The prepared capsules were administered to beagle dogs in
the morning with 60 ml of tap water by an oral gavage. After dosing,
the dogs were returned to metabolism cages with free access to water.
They were provided with standard food after the 12 hours sampling
time point.
A total of 5 ml of the whole blood samples were taken from
the jugular vein via 20 gauge needle of the dogs before and at the
following time points after dosing: 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16,
and 24 h. Plasma was obtained by centrifuging for 10 min at 4000
rpm, and was stored in a freezer at -20 0C before analysis.
The samples were extracted using the following procedure:
plasma (1.0 ml) was mixed with 50 μl oridonin methanol solution
(0.5 μg/ml) as an internal standard, and 6.0 ml ethyl acetate in a 10
ml plastic centrifuge tube, centrifuged at 4000 rpm for 10 min to
precipitate the proteins and the supernatant layer was evaporated in a
rotary centrifugal vacuum evaporator. The residue was reconstituted
in 100 μl mobile phase and 20 μl of the resulting solution was analyzed
by HPLC-UV method, as described in the section “Determination of
GLA content in phospholipid complex”.
Analysis of data:
The pharmacokinetic parameters were calculated by DAS 2.0
software. All values were expressed as the mean ± SD. Statistical
analysis was carried out using Student’s t-test. Significant difference
was regarded as p<0.05.
Results and Discussion
Preparation of GLA-PLC:
A good amount of aprotic organic solvents can be used as reaction
solvents to prepare phospholipid complex, for instance, methanol,
ethanol, tetrahydrofuran, acetone and so on [32,33]. In this study,
as a way to reduce the harm to human body or our environment,
more safety anhydrous ethanol was chosen as the solvent during the
process of GLA-PLC.
From the single factor experiment we can know that when the
reaction time further increased, there was no obviously difference
in the drug-phospholipid combination ratio (The combination ratio
were 31.2%, 62.4%, 72.8%, 91.3%, 91.5%, and 91.2% for 0.5 h, 1.0 h,
1.5 h, 2.0 h, 3.0 h, and 4.0 h, respectively.). On the other hand, due to
the thermal stability of phospholipid, when the reaction temperature
rose over 60 0C, the combination ratio was declined. Moreover,
the combination ratio was declined when the GLA concentration
exceeded 10 mg/ml. This phenomenon was due to the excessively high
concentration of the two material in the system, which may result
in insufficient contact of GLA with phospholipid in the solvent. The
combination ratio was increased with the increase of phospholipid
proportion. The experimental data were 45.6%, 85.8%, 89.4%, and
91.3% for GLA and phospholipid in 1:0.5, 1:1, 1:2, and 1:3 molar
ratios, respectively.
The optimal formulation and processing parameters to obtain the
maximum combination ratio of GLA-PLC were selected by orthogonal
experiment design. Theoretically, the optimum conditions for the
preparation of GLA-PLC were A2B3C3D3.The results are shown in
Table 2. In order to validate it, three batches were performed using the optimized preparation conditions (reaction temperature 40 0C,
reaction time 3 h, concentration 15 mg/ml, GLA: phospholipid 1:2).
The combination ratio of each batch of GLA-PLC was 94.6%, 95.3%,
and 95.0%, respectively.
Characterization of GLA-PLC:
Differential scanning calorimetry (DSC): DSC is a fast and
reliable thermo-analytical technique to obtain distinct information on
the polymorphism and crystallinity of the molecular interaction based
on a change in the sharp of peak. Figure 2 presents the DSC curves
of GLA (A), phospholipid (B), GLA-PPM (C) and GLA-PLC (D).
As shown in Figure 2, GLA showed one sharp endothermic peaks at
223 0C, suggested that it’s crystalline state. The phospholipid displays
a broad endotherm at 860 corresponding to its phase transition.
GLA-PPM exhibited two peaks at 86 0C and 223 0C corresponding
to phospholipid and GLA, respectively. However, the endothermic
peaks at 223 0C was disappeared in the DSC of GLA-PLC (Figure 2D), demonstrating the occurrence of molecular interactions between
GLA and phospholipid. These results indicated that there was no
crystallization of GLA in the GLA-PLC, GLA may be dispersed
molecularly or amorphously in the phospholipid molecule.
Powder X-ray diffraction (PXRD):
Figure 3 showed the powder
X-ray diffraction patterns of GLA (A), phospholipid (B), GLA-PPM
(C) and GAL-PLC (D). The spectrum of GLA exhibited numerous
sharp diffraction peaks, indicating that the GLA was present as
a crystalline pattern. No peaks were detected in the spectrum of
phospholipid, which illustrated its amorphous nature. The diffraction
pattern of the GLA-PPM revealed the characteristic peaks of
GLA, suggested that simple physical mixing had no effect on the
interaction between GLA and phospholipid. On the other hand, all
the crystalline peaks had disappeared in GLA-PLC, indicated that
GLA in a phospholipid matrix was either molecularly dispersed or
in an amorphous form [34], which was consistent with the result of
the DSC.
Scanning electron microscopy (SEM):
The representative
surface morphology of GLA (A), phospholipid (B), GLA-PPM (C)
and GLA-PLC (D) are presented in (Figure 4). GLA appeared as
smooth-surfaced rectangular crystalline structures with different
size and the similar crystal structure can be observed in the physical
mixture. For GLA-PLC, the morphology was changed completely
and rectangular crystalline structures were not observed, indicating
that GLA was transformed into an amorphous state.
Solubility studies:
From the data in Table 3, we can conclude that the solubility of
GLA-PLC in water and n-octanol increased significantly (P < 0.01)
in comparison with GLA. The solubility of the GLA-PLC and GLAPP
Min water was about 13.7 times and 1.6 times higher than GLA. This improvement in solubility could be explained by the amorphous
form of GLA and the amphipathic nature and solubilizing effect of
phospholipid. In addition, the date illustrated that the solubility of
GLA-PLC in n-octanol was slightly increased than GLA, which in
turn might result in improved absorption across the gastrointestinal
tract. In short, the solubility of GLA-PLC increased in water and
n-octanol means that the drug could be well dissolved and absorbed
in gastrointestinal tract.
Preparation and <em>in vitro</em> drug release of GLA-PLC sustainedrelease pellets:
Enhancement of the drug solubility in the inner compartment
aided the control of the drug release by the outer rate-controlling
membrane onto the pellets was the main technical route in this
article. In order to design the reservoir-type sustained release pellets,
the poorly water soluble GLA was prepared into GLA-PLC which
were used as the core material for the layering process. For facilitate
the preparation of pellets, lactose was added to GLA-PLC to get freeflowing
powder, due to the viscous property of phospholipid. Talc
was used as lubricants to prevent aggregation between the pellets and
improve powder flow properties. The pellets core were prepared by
extrusion-spheronization method, which is one of the conventional
methods used since years in pharmaceutical industry [35,36].
Enhancement of the drug solubility in the inner compartment
aided the control of the drug release by the outer rate-controlling
membrane onto the pellets was the main technical route in this
article. In order to design the reservoir-type sustained release pellets,
the poorly water soluble GLA was prepared into GLA-PLC which
were used as the core material for the layering process. For facilitate
the preparation of pellets, lactose was added to GLA-PLC to get freeflowing
powder, due to the viscous property of phospholipid. Talc
was used as lubricants to prevent aggregation between the pellets and
improve powder flow properties. The pellets core were prepared by
extrusion-spheronization method, which is one of the conventional
methods used since years in pharmaceutical industry [35,36].
Pharmacokinetic studies in dogs:
The plasma concentration-time profiles of GLA in beagle dogs
following the oral administration of GLA, GLA-PLC, and GLA-PLC
sustained release pellets are presented in (Figure 6), and the major
pharmacokinetic parameters are summarized in Table 4. From
the data we can know that the plasma concentration of GLA from
GLA-PLC exhibited a higher Cmax (218.2 ± 36.3 ng/ml, 1.85-fold) in
comparison with free GLA (118.2 ± 25.2 ng/ml). AUC(0-∞) of GLA in
GLA-PLC (1567.4 ± 146.3 ng/ml h) was significantly higher than that
of the free GLA (716.7 ± 108.2 ng/ml h), with 2.19-fold increased oral
bioavailability.
Additionally, the maximum plasma concentration obtained from
GLA-PLC sustained-release pellets were reach at 4.5 ± 1.0 h and
decreased more slowly than that of free GLA and GLA-PLC capsule.
The AUC(0-∞) of GLA-PLC sustained release pellets were 1485.6 ±
104.8 ng/ml.h (2.07-fold), in contrast to 716.7 ± 108.2 ng/ml h of free
GLA. In the present case, enhanced oral bioavailability of GLA was
achieved by phospholipid complex with the relative bioavailability of
GLA-PLC and GLA-PLC sustained-release pellets were 218.7% and
207.3% compared with the free GLA, respectively.
Pharmacokinetics findings demonstrated that GLA-PLC and
GLA-PLC sustained-release pellets showed better absorption rate,
and/or sustained release behavior in vivo than free GLA. This may be
attributed to the following reasons:
(1) Compared to crystalline state of GLA, an amorphous state
of GLA in GLA-PLC had better solubility and in vitro drug release
profiles of GLA;
(2) Phospholipids being an important component of cell
membrane to maintain the fluidity of cell membrane and owing to
the amphiphilic nature, the GLA-PLC are considered to penetrate the
cell membrane and enter the cytoplasm of living mammalian cells
without disturbing the cellular lipid bilayers;
(3)The combination of GLA to phospholipids can enhance the
absorption of GLA through lymphatic pathway and may reduce the
exposure of the GLA in the gastrointestinal tract and protect the GLA
from degradation by the enzymes [37,38].
(4) The coating layer formed a good barrier membrane to control
the drug release from the pellets.
Conclusion
In this paper, GLA-PLC and GLA-PLC sustained-release pellets
were successfully prepared for the first time. The physicochemical
characteristics of GLA-PLC were indicated by DSC, PXRD, and
SEM techniques and the studies showed that GLA in a phospholipid
matrix was either molecularly dispersed or in an amorphous form.
Our research based on solubility and in vitro drug release studies
clearly demonstrated a higher solubility and complete drug release
profiles for GLA-PLC than free GLA. Besides that, GLA-PLC
sustained-release pellets exhibited sustained release characteristics
with over 95% total release in 12 h. The pharmacokinetic studies in
beagle dogs demonstrated that GLA-PLC and GLA-PLC sustainedrelease
pellets showed 2.19-fold and 2.07-fold improvement than
the free GLA by oral dosage in beagle dogs. Further, enhanced oral
bioavailability, steady plasma concentration, and prolonger Tmax were
simultaneously obtained from GLA-PLC sustained release pellets,
indicating a promising way to achieve the optimal oral bioavailability
with sustained release property for poorly water soluble drugs.
References
Citation
Miao YF, Sun JQ. Preparation and Evaluation of Glaucocalyxin A Sustained-Release Pellets Based on Phospholipid Complex System with Enhanced Bioavailability. J Pharmaceu Pharmacol. 2020; 8(1): 7.