
Journal of Surgery
Download PDF
Review Article
*Address for Correspondence: David M. Jablons, MD, Thoracic Oncology Laboratory, Department of Surgery, University of California San Francisco, San Francisco, CA, USA, E-mail: David.Jablons@ucsfmedctr.org
Il-Jin Kim, PhD, Thoracic Oncology Laboratory, Department of Surgery, University of California San Francisco, San Francisco, CA, USA, E-mail: kimij@cc.ucsf.edu
Citation: Kim HK, Jablons DM, Kim IJ. Multistep Progression from Atypical Adenomatous Hyperplasia to Lung Adenocarcinoma: Clinico-Pathologic, Epigenetic and Genetic Aspects. J Surgery. 2013;1(1): 10.
Copyright © 2013 Kim HK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Surgery | ISSN: 2332-4139 | Volume: 1, Issue: 1
Submission: 03 July 2013 | Accepted: 07 August 2013 | Published: 12 August 2013
 Prior to the introduction of such an ‘attractive’ high-resolution CT scanning, Noguchi et al. first conducted a large systematic study regarding the histologic features of small peripheral lung ADC measuring 2 cm or less in diameter and then classified them into six subtypes [34] Type A corresponds to localized BAC; type B, localized BAC with foci of collapsed alveolar structures; type C, localized BAC with foci of active fibroblastic proliferation; type D, poorly differentiated ADC; type E, tubular ADC; and type F, papillary ADC with evidence of compressive and destructive growth. Types A, B, and C (“replacement” early ADC), unlike types D, E, and F (“nonreplacement” invasive ADC), represent a distinct group, because they show a lepidic growth pattern, which is characterized by tumor cell growth replacing normal alveolar lining cells. These tumors with replacement or lepidic growth pattern appear radiographically as a localized GGO lesion at high-resolution CT scans.
Prior to the introduction of such an ‘attractive’ high-resolution CT scanning, Noguchi et al. first conducted a large systematic study regarding the histologic features of small peripheral lung ADC measuring 2 cm or less in diameter and then classified them into six subtypes [34] Type A corresponds to localized BAC; type B, localized BAC with foci of collapsed alveolar structures; type C, localized BAC with foci of active fibroblastic proliferation; type D, poorly differentiated ADC; type E, tubular ADC; and type F, papillary ADC with evidence of compressive and destructive growth. Types A, B, and C (“replacement” early ADC), unlike types D, E, and F (“nonreplacement” invasive ADC), represent a distinct group, because they show a lepidic growth pattern, which is characterized by tumor cell growth replacing normal alveolar lining cells. These tumors with replacement or lepidic growth pattern appear radiographically as a localized GGO lesion at high-resolution CT scans.
 However, if a GGO nodule (pathologically AAH or BAC) is not surgically resected and merely followed up by repeated CT scans without treatment, it increases in size and a solid component within the lesion tends to appear and increase in extent [46,47]. Takashima et al. conducted a serial CT follow-up study on the natural course of lung ADCs with GGO components and showed that GGO subsequently increased in size in 75% of patients, 17% of patients developed solid components within the nodule, and the solid portions increased in 23% patients [46]. Such a GGO nodule containing the solid component in it is called mixed GGO (partsolid nodule), whereas a GGO nodule without any solid component is called pure GGO (non-solid nodule) [32,48] (Figure 1). Solid components within GGO nodules pathologically represent areas of collapsed alveoli or fibroblastic proliferation and these mixed GGO nodules correspond to Noguchi type B and C lesions [34,46,49,50]. Greater the solid component corresponds with greater possibility of an invasive growth component [51-53]. Kodama et al. demonstrated that radiologic GGO components correlated with BAC components in pathologic specimens of ADC [54]. In the same context, AAH and BAC are manifested by pure GGO, whereas invasive ADCs include a greater solid component within GGO on CT scans. There have been several reports that lung cancer patients with smaller solid component had a much better prognosis than those with greater solid component [39,54,55]. For this reason, the diagnosis of BAC is based upon a premise that the entire tumor has no areas of invasion upon pathologic examination. It is therefore highly controversial to make a final diagnosis of BAC based on a small biopsy or cytology specimen alone. However, it should be noted that identification of small GGO nodules at CT in fact may lead to overdiagnosis and unnecessary treatment [56]. The question can be raised whether surgical resection is really needed for such a small pure GGO lesion, considering the fact that it grows very slowly or not at all during follow-up when untreated. In the same context, many surgeons demonstrated that limited surgical resection such as wedge resection rather than lobectomy is enough for small pure GGO lesions [57-59].
However, if a GGO nodule (pathologically AAH or BAC) is not surgically resected and merely followed up by repeated CT scans without treatment, it increases in size and a solid component within the lesion tends to appear and increase in extent [46,47]. Takashima et al. conducted a serial CT follow-up study on the natural course of lung ADCs with GGO components and showed that GGO subsequently increased in size in 75% of patients, 17% of patients developed solid components within the nodule, and the solid portions increased in 23% patients [46]. Such a GGO nodule containing the solid component in it is called mixed GGO (partsolid nodule), whereas a GGO nodule without any solid component is called pure GGO (non-solid nodule) [32,48] (Figure 1). Solid components within GGO nodules pathologically represent areas of collapsed alveoli or fibroblastic proliferation and these mixed GGO nodules correspond to Noguchi type B and C lesions [34,46,49,50]. Greater the solid component corresponds with greater possibility of an invasive growth component [51-53]. Kodama et al. demonstrated that radiologic GGO components correlated with BAC components in pathologic specimens of ADC [54]. In the same context, AAH and BAC are manifested by pure GGO, whereas invasive ADCs include a greater solid component within GGO on CT scans. There have been several reports that lung cancer patients with smaller solid component had a much better prognosis than those with greater solid component [39,54,55]. For this reason, the diagnosis of BAC is based upon a premise that the entire tumor has no areas of invasion upon pathologic examination. It is therefore highly controversial to make a final diagnosis of BAC based on a small biopsy or cytology specimen alone. However, it should be noted that identification of small GGO nodules at CT in fact may lead to overdiagnosis and unnecessary treatment [56]. The question can be raised whether surgical resection is really needed for such a small pure GGO lesion, considering the fact that it grows very slowly or not at all during follow-up when untreated. In the same context, many surgeons demonstrated that limited surgical resection such as wedge resection rather than lobectomy is enough for small pure GGO lesions [57-59].

In contrast, the incidence of EGFR mutations in invasive ADC ranged from 23% to 50%, which seems to be relatively higher than that of AAHs [77,90,91,103]. Sakuma et al. reported that 15 of 17 (88%) pure nonmucinous BAC (AIS) and 49 of 65 (75%) invasive ADC with a nonmucinous BAC component had EGFR mutations [92]. These results imply that persistent EGFR signaling from activating EGFR mutations would be essential for the development and maintenance in lung ADCs with a nonmucinous BAC component [92]. Apart from these dissimilar frequencies of EGFR mutations based on the invasiveness of lung ADCs, the findings that EGFR mutations are detected in noninvasive tumors as well as invasive tumors indicate that EGFR mutation is an early event in the pathogenesis of lung ADCs [70,104-106].
Multistep Progression from Atypical Adenomatous Hyperplasia to Lung Adenocarcinoma: Clinico-Pathologic, Epigenetic and Genetic Aspects
Hong Kwan Kim1,3, David M. Jablons1,2* and Il-Jin Kim1,2*
- 1Thoracic Oncology Laboratory, Department of Surgery, University of California San Francisco, San Francisco, CA, USA
- 2Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA, USA
- 3Department of Thoracic and Cardiovascular Surgery, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
*Address for Correspondence: David M. Jablons, MD, Thoracic Oncology Laboratory, Department of Surgery, University of California San Francisco, San Francisco, CA, USA, E-mail: David.Jablons@ucsfmedctr.org
Il-Jin Kim, PhD, Thoracic Oncology Laboratory, Department of Surgery, University of California San Francisco, San Francisco, CA, USA, E-mail: kimij@cc.ucsf.edu
Citation: Kim HK, Jablons DM, Kim IJ. Multistep Progression from Atypical Adenomatous Hyperplasia to Lung Adenocarcinoma: Clinico-Pathologic, Epigenetic and Genetic Aspects. J Surgery. 2013;1(1): 10.
Copyright © 2013 Kim HK, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Surgery | ISSN: 2332-4139 | Volume: 1, Issue: 1
Submission: 03 July 2013 | Accepted: 07 August 2013 | Published: 12 August 2013
Abstract
Detection of small peripheral ground-glass opacity nodules has increased due to the advances in imaging modalities and the widespread use of computed tomography screening. Pathologic examination of these nodules revealed that they have a pure lepidic or replacement growth pattern such as atypical adenomatous hyperplasia or adenocarcinoma in situ (formerly known as bronchioloalveolar carcinoma). When untreated, ground-glass opacity nodules gradually develop a solid component. The greater the solid component or the invasive component, the less favorable outcomes after treatment for patients with ground-glass opacity nodules. Based on the clinical, radiologic and pathologic findings, the concept of multistep progression from preinvasive atypical adenomatous hyperplasia through noninvasive adenocarcinoma in situ to invasive adenocarcinoma has been postulated. Recently, evidence has accumulated explaining this putative concept by molecular alterations, including activating mutation of oncogenes and inactivation of tumor suppressor genes by epigenetic changes or loss of heterozygosity. This review 1) comprehensively outlines the accumulated knowledge regarding radiologic and pathologic features of adenocarcinoma and its precursor which presents as ground-glass opacity and 2) summarizes the molecular basis of the multistep progression to lung adenocarcinoma. As a result, we believe identification of undiscovered molecular markers involved in the progression of lung adenocarcinoma is critical for early detection of lung cancer and the development of targeted therapeutic and chemoprevention strategies.Keywords
Atypical adenomatous hyperplasia; Adenocarcinoma in situ; Invasive adenocarcinoma; Ground-glass opacity; Molecular alterationsAbbreviations
AAH: Atypical Adenomatous Hyperplasia; ADC: Adenocarcinoma; AIS: Adenocarcinoma in situ; BAC: Bronchioloalveolar Carcinoma; CT: Computed Tomography; GGO: Ground-Glass Opacity; LOH: Loss of Heterozygosity; MIA: Minimally Invasive Adenocarcinoma; NGS: Next Generation Sequencing; NSCLC: Non-Small Cell Lung Cancer; PET: Positron Emission Tomography; WHO: World Health OrganizationIntroduction
Lung cancer is the leading cause of cancer deaths in the United States and worldwide with over 1.3 million deaths in 2008 [1-3]. Despite the fact that enormous resources have been spent on research involving molecular and therapeutic aspects of lung adenocarcinoma (ADC), there has been no significant improvement in the mortality associated with lung cancer for the past 25 years. This can be attributed in part to untimely diagnosis at advanced stages or recurrence occurring even after optimal treatment at early stages. About 70% of patients are diagnosed with lung cancer at advanced stages when there is little chance to cure [4]. Although patients diagnosed at early stages receive curative-intent complete resection by surgery, about 20% of them will not survive due to recurrence within 5 years [5-12]. One cause may be that patients already have microscopic systemic metastases in other distant organs at the time of surgery. In order to reduce the mortality and eventually to overcome lung cancer, understanding carcinogenesis and tumor progression is paramount.ADC is the most common histologic type of non-small cell lung cancer (NSCLC) in the United States, accounting for almost half of all lung cancers [13,14]. ADC tends to develop distant organ metastases easily even in early stage compared to squamous cell carcinoma, underscoring the need for early detection methods. A multistep progression concept from a precursor lesion (atypical adenomatous hyperplasia, AAH) through noninvasive tumor (bronchioloalveolar carcinoma, BAC; BAC is currently renamed adenocarcinoma in situ (AIS), but the term BAC from published articles remained unchanged in this review) to invasive ADC has been postulated based on a variety of clinical, pathologic, and molecular studies [15-18]. It was reported that AAH was frequently detected at the periphery of invasive ADC in surgically resected lungs for pulmonary carcinoma [17,19]. Other reports showed the multistep progression of lung ADC with ground-glass opacity (GGO) features by performing a long-term follow-up with regular CT scans in the same patient [20,21]. This concept appears to be consistent with radiologic-pathologic features and molecular events.
A clear understanding of biological events during the progression of lung ADC will be critical for identifying molecular biomarkers related to carcinogenesis and tumor progression. Ultimately, it will facilitate early detection of lung cancer, contribute the development of targeted therapeutic strategies, and ideally widen the scope of chemoprevention. This review comprehensively summarizes the accumulated knowledge regarding (1) radiologic features of the preinvasive lesion or early-stage ADC, (2) pathologic findings of AAH, BAC (AIS) or ADC, and (3) genetic and (4) epigenetic alterations associated with the concept of multistep progression of ADC to provide a better understanding of carcinogenesis in a subset of lung ADC.
Radiologic and Pathologic Features
With recent advances in diagnostic imaging modalities, small indeterminate nodules such as GGO lesions have been increasingly detected on high-resolution computed tomography (CT) scans [22-25]. In addition, the introduction of low-dose helical CT scanning for lung cancer screening has further increased the detection rate of small GGO lesions. Recently, the observation from the National Lung Screening Trial that low-dose CT screening can reduce mortality from lung cancer obviously justified the use of CT screening in clinical practices and it is anticipated that GGO lesions will be detected more commonly [26].GGO is defined as hazy opacity with increased lung attenuation that does not obscure underlying bronchial structures or pulmonary vessels at high-resolution CT [27] (Figure 1). Since GGO only refers to a morphologic finding on CT scans, nodules with GGO morphology may represent various pathologic entities such as inflammation, alveolar hemorrhage, eosinophilic lung disease, pulmonary lymphoproliferative disorder, organizing pneumonia, and neoplasm [28]. When GGO nodules are transiently observed and disappear thereafter, they are more likely to have been inflammation or hemorrhage. In contrast, GGO nodules that persist for more than several months could be a sign of early-stage ADC or its precursor [29-35]. Nakata et al. reported that GGO nodules persistently observed for several months turned out to be early-stage ADC such as BAC (AIS) or AAH [29]. The appeal of predicting the pathologic diagnoses based on noninvasive tests like CT scans led numerous attempts to elucidate the histopathologic findings of GGO nodules and correlate them with CT findings [29,31-33].
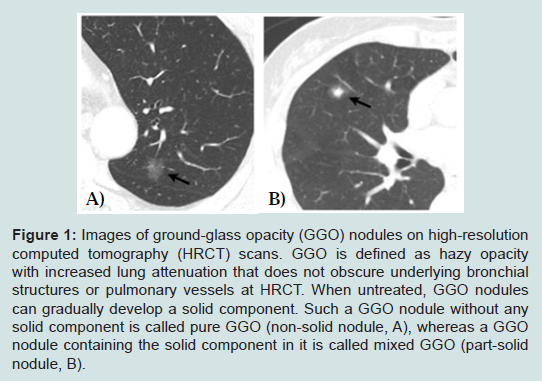
Figure 1: Images of ground-glass opacity (GGO) nodules on high-resolution computed tomography (HRCT) scans. GGO is defined as hazy opacity with increased lung attenuation that does not obscure underlying bronchial structures or pulmonary vessels at HRCT. When untreated, GGO nodules can gradually develop a solid component. Such a GGO nodule without any solid component is called pure GGO (non-solid nodule, A), whereas a GGO nodule containing the solid component in it is called mixed GGO (part-solid nodule, B).
Since Noguchi et al. first proposed this novel classification, significant changes have been made in the World Health Organization (WHO) classification of lung ADC, which reflects our better understanding of the histopathologic features [36]. A major change made in the 1999 WHO classification and maintained in the 2004 WHO classification was the addition of AAH as a premalignant lesion [36]. AAH is defined as a localized proliferation of mildly to moderately atypical type II pneumocytes and/or Clara cells lining alveolar walls and sometimes, respiratory bronchioles when underlying interstitial inflammation and fibrosis are absent (Figure 2). The proliferation results in focal lesions of the peripheral lung, usually less than 5 mm in diameter [37]. On the other hand, BAC is a localized noninvasive ADC with a pure lepidic growth pattern restricted to neoplastic cells along preexisting alveolar structures and no evidence of stromal, pleural, or vascular invasion [36,38] (Figure 2). By definition, BAC corresponds to Noguchi type A and B lesions. Numerous clinical studies on solitary small (< 2 or 3cm) lung ADC with a pure lepidic growth, that is, BAC have shown 100% diseasefree survival after a curative-intent surgical resection [34,39-45].
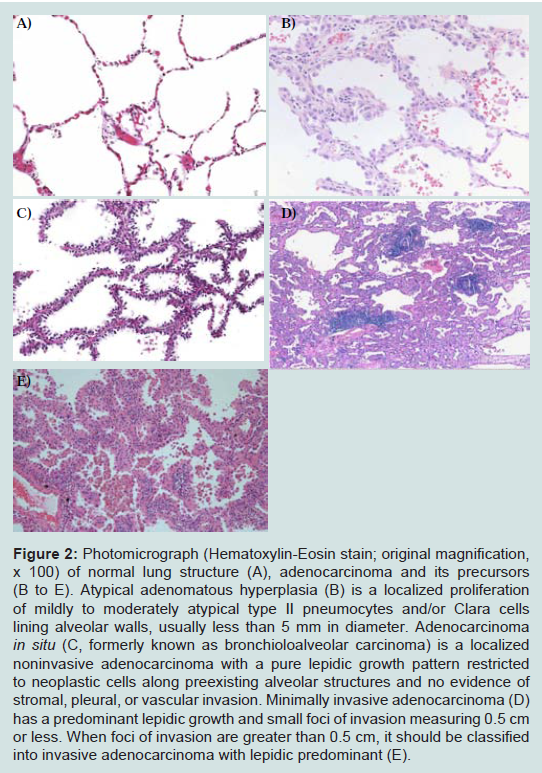
Figure 2: Photomicrograph (Hematoxylin-Eosin stain; original magnification, x 100) of normal lung structure (A), adenocarcinoma and its precursors (B to E). Atypical adenomatous hyperplasia (B) is a localized proliferation of mildly to moderately atypical type II pneumocytes and/or Clara cells lining alveolar walls, usually less than 5 mm in diameter. Adenocarcinoma in situ (C, formerly known as bronchioloalveolar carcinoma) is a localized noninvasive adenocarcinoma with a pure lepidic growth pattern restricted to neoplastic cells along preexisting alveolar structures and no evidence of stromal, pleural, or vascular invasion. Minimally invasive adenocarcinoma (D) has a predominant lepidic growth and small foci of invasion measuring 0.5 cm or less. When foci of invasion are greater than 0.5 cm, it should be classified into invasive adenocarcinoma with lepidic predominant (E).
Nonetheless, many researchers and clinicians still use the term BAC for a broad spectrum of tumors from solitary small noninvasive lung tumors with a 100% 5-year survival [34] to invasive ADCs with minimal invasion that also have almost 100% 5-year survival [60,61] or mixed subtype invasive ADCs [62,63], creating great confusion in both clinical and research fields. Recently, new lung ADC classifications have been proposed by the European Respiratory Society, International Association for the Study of Lung Cancer, and American Thoracic Society [18]. The proposal strongly recommended discontinuing the use of the term BAC and instead recommended that the term ‘AIS’ should be used for small (≤3 cm), solitary ADCs with pure lepidic growth to define patients who should have 100% disease-specific survival upon complete resection. When small (≤3 cm), solitary ADCs have predominant lepidic growth and small foci of invasion measuring 0.5 cm or less, a new concept of ‘minimally invasive adenocarcinoma (MIA)’ is proposed to define patients who should have nearly 100% disease-specific survival upon complete resection. For invasive ADCs, comprehensive histological subtyping should be used to assess histologic patterns semiquantitatively in 5% increments, choosing a single predominant pattern.
Concept of Multistep Progression
Based on clinical implications and pathologic findings, many researchers proposed a hypothesis of multistep carcinogenesis in which some lung ADCs arise from preneoplastic lesions called AAH, which progress to AIS, eventually developing into invasive ADC [15-18]. This concept was initially proposed in the field of colorectal cancer, in which colorectal carcinogenesis involves a multistep process from normal mucosa and inflammation, through early and late adenomas to invasive carcinoma [64,65]. If specimens for histologic analysis could easily be obtained repeatedly over time in the same patient, it would be an ideal method to elucidate the natural history of cancer. In colon cancer, this longitudinal study can be performed because it is relatively easy to obtain colon cancer specimens via an endoscopic approach. However, in lung cancer, such a procedure is more challenging especially in peripheral lung cancer, because it is rarely accessible via an endoscopic approach. Furthermore, it is much more difficult to obtain specimens if the lesion of interest is at an early stage such as AAH [66]. Although serial morphologic changes in imaging tests such as CT or positron emission tomography (PET) can be an alternative way to demonstrate the multistep progression of lung ADC [20,21], it cannot be guaranteed that radiographic features are equivalent to pathologic findings. Otherwise, it cannot help but compare individual lesions from different patients and a temporal assumption is inevitable in this cross-sectional method, which should be interpreted with caution. It should also be noted that the time point at which genetic or epigenetic alterations occur might differ from patient to patient, since it is still difficult to tell exactly when these alterations first occur. In addition, genes affected in a specific pathway might vary from tumor to tumor, because not all molecular changes will be fully penetrant and only a portion of tumors will gain a specific molecular change [67]. Furthermore, since lung ADC can no longer be considered a single type of tumor but rather a group of distinct subsets that arise from different molecular pathways, the concept of multistep progression carcinogenesis is not necessarily applicable to all ADCs [68].In spite of all these limitations regarding the concept of multistep progression of lung ADC, there is sufficient pathologic and molecular evidence to substantiate this putative hypothesis of multistep carcinogenesis in lung ADC. First, AAH is frequently detected at the periphery of invasive ADC in lung cancer patients [17,19]. This phenomenon is relevant to the histopathological similarity between AAH and AIS [69-71]. Second, numerous molecular studies demonstrate that specific genetic alterations occur at similar frequencies in AAH and ADC, which reinforce the concept of AAH-AIS-ADC progression [66]. Multiple studies demonstrated close association between AAH and lung ADC including clonality [72,73], K-ras mutations [74,75], K-ras polymorphisms [76], epidermal growth factor receptor (EGFR) mutations [77], p53 expressions [78], loss of heterozygosities [79], methylations [80], telomerase overexpressions [81], eukaryotic initiation factor 4E expressions [82], epigenetic alterations in the Wnt pathway [83], and FHIT expressions [84]. For example, activating K-ras mutations are found with similar frequency in AAH and ADC [75,85] and the mutually exclusive features of EGFR and K-ras mutations observed in lung ADC are also found in AAH [74]. In addition, loss of heterozygosity events at specific chromosomal regions are detected at similar frequencies in AAH and ADC [79,86]. Third, the postulated progression of AAH to AIS and then ADC is supported by conditional oncogenic mouse models in which EGFR or K-ras genes are activated. In both types of mice, AAH-like lesions are detected before ADCs develop, implying AAH as a precursor of ADC [87,88].
Genetic Alteration
Lung ADC arises from the accumulation of enormous genetic and epigenetic changes, giving advantages to neoplastic cells in cellular growth and/or survival with progression depending mainly on the balance between oncogene activation and tumor suppressor gene inactivation [89]. To date, over 100 oncogenes have been identified such as ras and tyrosine kinase receptors (EGFR, c-erb-B2 (HER-2/neu)). Many of these behave dominatly in that only one allele needs to be overexpressed to have effect [77]. In contrast, tumor suppressor genes behave as recessive genes and thus both alleles need to be inactivated either by epigenetic modifications (predominantly by promoter methylation), allelic deletion or mutation [71].EGFR
Many AAH and AIS cases have been found to harbor EGFR gene mutation and these findings suggest that EGFR mutation has a critical role in the pathogenesis of lung ADC [75,77,90-93]. Furthermore, transgenic mouse models expressing mutated EGFR genes in type II pneumocytes develop AAH, BAC, and invasive ADC with a nonmucinous BAC component in the lung in a time-dependent manner [87,94]. Ligand binding to EGFR leads to receptor tyrosine kinase activation and a series of downstream signaling activation mediates increased cellular proliferation, migration, invasion and suppression of apoptosis [95]. Mutations responsible for its oncogenic activation and depending on the ligand (EGF, transforming growth factor-alpha, insulin-like growth factor-1, platelet-derived growth factor, amphiregulin) favor deregulated proliferation, differentiation, apoptosis and angiogenesis [71]. Somatic mutations of EGFR are characterized by two major hotspots, in-frame deletions in exon 19 and a specific missense mutation in exon 21 (ie, L858R), which constitute almost 90% of the EGFR mutations in lung ADC [90-95]. ADCs with these mutations have been reported in certain demographic populations, including female gender, never or light smoking status and East Asian ethnicity [101,102]. Histologically, they tend to be associated with ADC especially with nonmucinous BAC component [101,102]. More Importantly, patients with lung ADC harboring these mutations are responsive to EGFR tyrosine kinase inhibitors such as gefitinib and erlotinib [96-101].
Based on these clinical circumstances, many investigators tried to elucidate the evidence of multistep progression from noninvasive to invasive tumors by comparing the incidence of EGFR mutation between AAH and AIS or MIA. The incidence of EGFR mutations in AAH varies from 3% to 44% [77,90,103]. Kozuki et al. reported that EGFR mutations were detected in 44% of AAHs (four of nine cases) [103], whereas Yoshida et al. showed that the incidence of EGFR mutation in AAH was only 3% (one out of 35 cases) [77]. The differences in these frequencies might be due to the sensitivity of the assays to detect EGFR mutations as there were fewer AAH cells than in the surrounding normal cells. Although the refinement of microdissection techniques coupled with improvements in PCR amplification has made it possible to study AAH lesions at the molecular genetic level [19], it remains technically challenging to conduct a molecular analysis for AAHs due to their small size. Nonetheless, the fact that EGFR mutations are detected even in AAH is undeniable and this suggests that EGFR mutations occur early in the development of lung ADC before progression to invasive tumors (Figure 3).
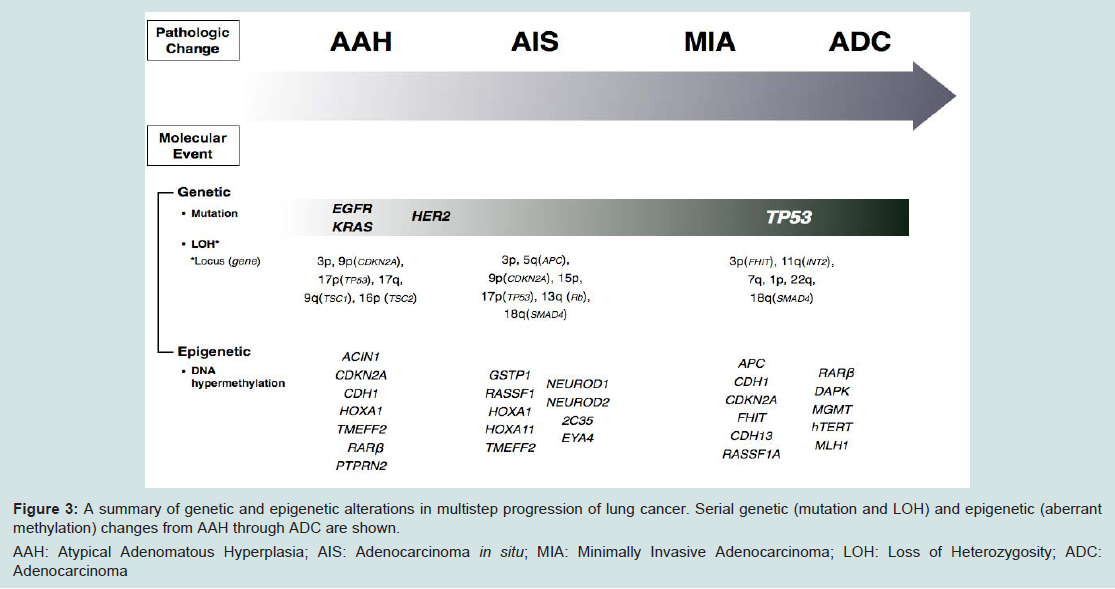
Figure 3: A summary of genetic and epigenetic alterations in multistep progression of lung cancer. Serial genetic (mutation and LOH) and epigenetic (aberrant methylation) changes from AAH through ADC are shown.
AAH: Atypical Adenomatous Hyperplasia; AIS: Adenocarcinoma in situ; MIA: Minimally Invasive Adenocarcinoma; LOH: Loss of Heterozygosity; ADC: Adenocarcinoma
While many researchers reported that EGFR mutation is an early event during the multistep progression of lung ADCs, EGFR amplification is considered to be a late event. Soh et al. found that EGFR mutations were present in 39.5% of noninvasive tumors and 50% of invasive tumors, while EGFR copy number was increased in 7.9% and 31.8% of noninvasive and invasive tumors, respectively [70]. They concluded that EGFR mutations occur early during the multistage pathogenesis of lung ADCs, but increased EGFR copy number is a late event during tumor development and plays a role in the progression of lung ADC independent of the initiating molecular events. This finding is in line with other reports that demonstrate EGFR mutations preceding amplification, with amplification favoring the mutant allele [107].
Like EGFR mutations, c-erb-B2 mutations are found most commonly in female Asian non-smokers with adenocarcinoma and are mutually exclusive with KRAS mutations [108]. HER2 protein expression was reported in 7% of AAH whereas up to 67% of lung ADC showed positive staining for HER2 protein [109,110]. HER2 overexpression in AAH and BAC suggests that the tyrosine kinase signaling pathway is altered by a variety of mechanisms before adenocarcinoma reaches its invasive stage. HER2 protein staining is also related to increased cellularity and pleomorphism of AAH. Therefore, it has been speculated that AAH is premalignant and abnormal c-erb-B2 proto-oncogene expression may occur in the later carcinogenic sequence [109].
K-rasK-ras is a member of the ras family of oncogenes, and is located downstream of the EGFR surface receptor pathway. K-ras gene encodes a membrane-associated guanine nucleotide-binding protein of approximately 21 kD in size, designated p21-ras and activation of K-ras genes by mutations may contribute to malignant transformation [111-115]. K-ras mutations are found in 10 to 30% in NSCLC, especially ADCs, and over 90% of the mutations occurred in codon 12, occasionally in codon 13, and rarely in codon 61 [116-120]. Like EGFR mutation, K-ras mutation has also been detected in certain demographic populations, including non-East Asian male and ever smokers. Histologically, tumors tend to be associated with wild-type EGFR and contain mucinous BAC component features [102,117,118,121-123]. More importantly, K-ras mutations are found more frequently in patients who are resistant to therapy with EGFR tyrosine kinase inhibitors [124].
Initial studies mainly focused on the K-ras proto-oncogene as a likely target of mutational activation for several reasons [19]: (1) activating mutations of K-ras are detected in 24-50% of lung ADCs, but they are much less frequent in other subtypes of lung cancer [117,125]; (2) K-ras mutations are easy to detect because they involve straightforward base substitutions that predictably target codon 12 [117]; and (3) models of lung tumorigenesis addressing the chronological sequence of mutational events predict that K-ras activation is an early event that precedes malignant growth [85,126,127]. Therefore, K-ras mutations are the most extensively investigated genetic alterations in AAH and previous reports demonstrate that K-ras mutations are found in 15-50% of AAH [15,75,93,128,129]. These findings indicate that K-ras mutation is also an early event in the multistep carcinogenesis of lung ADC [15,75,93,128] (Figure 3).
Although K-ras mutation is frequently found in AAH, its incidence is relatively less frequent than that of EGFR mutations in AAH. K-ras mutations in AAH or in ADCs with a nonmucinous BAC feature were much less frequent than those with a mucinous BAC component [130]. Moreover, AAHs are strikingly similar to nonmucinous BAC, but quite different from mucinous BAC in histopathologic features. These observations suggest that AAH do progress sequentially to nonmucinous BAC, but not to mucinous BAC [130].
TP53
Mutation of the TP53 gene is one of the most frequent genetic alterations in lung cancer. TP53 is a tumor suppressor gene and its protein product is considered to play an important role in the control of cell cycle, DNA repair, apoptosis, and cell differentiation [131]. It reduces Rb phosphorylation and induces a G1-S checkpoint arrest to allow DNA repair or to drive cells to apoptosis mediated by Bax/Bcl-2 [71]. Its funtion is lost by mutation or inhibition of p53 pathway. Abnormalities of the TP53 gene, mainly missense mutations, result in an impairment of the normal functions of the TP53 gene [131]. Since TP53 mutations lead to intranuclear accumulation of nonfunctional, stabilized p53 protein, immunohistochemical detection of p53 protein is an indirect measurement of TP53 gene mutations. Using this method, the expresion of p53 protein in AAH, BAC and ADC has been studied by several researchers. Kerr et al. demonstrated that p53 protein was detected in 28% and 53% of AAH and ADC, respectively [109]. Kitamura et al. showed p53 expression in 5% to 8% of AAH lesions and 8% to 62% of BAC [78]. These findings indicate that the frequency of p53 nuclear accumulation sequentially increases from AAH to BAC and ADC. Therefore, this suggests that p53 overexpression might not be common even in the earliest stage of lung ADC and thus might be related to invasivenss in the tumor progression of lung ADC rather than initiation of tumor.
Loss of heterozygosity (LOH)
Inactivation of tumor suppressor genes is also an important factor in lung carcinogenesis. Loss of function of a specific gene occurs mainly by aberrant DNA methylation of its promoter region (will be described in the next section) and/or loss of heterozygosity (LOH) of the chromosomal region on which the gene is located. Many reportsdemonstrate that tumor and adjacent normal tissue from lung cancer patients contain LOH at distint regions of chromosomes [132-136]. Deletions on chromosomal arms 3p, 2p, 12p, 9p, 8p, and 17p have been found to be widespread throughout the lungs of smokers even in the absence of overt histopathologic changes, suggesting that these alterations occur during the earliest stages of lung tumorigenesis [132-138].
LOH accumulates in crucial chromosomal regions in a stepwise manner during the multistep sequential progression of lung ADC [139]. When compared to histologically normal adjacent lung, AAH shows LOH of distinct regions of chromosomes 3p (18%), 9p (p16INK4a, 13%), 9q (53%), 16p, 17q, and 17p (TP53, 6%), and these are changes also frequently detected in lung ADCs [140-143]. A detailed investigation of loci related to LOH in BACs and small ADCs, showed localized loss of 5q(APC), 9p(CDKN2A), 13q(RB1), 17p(TP53) and 18q(SMAD4) in BAC, while LOH of 3p(FHIT) and 11q(INT2) did not become prominent until invasion [139]. This finding is consistent with the observed persistence of FHIT protein expression in AAH and BAC but loss of expression in invasive disease [84]. FHIT is a member of the histidine triad gene family involved in purine metabolism and it is known to work as a tumor suppressor gene. This late loss of FHIT is a very interesting finding in light of the observation of methylation of this locus in lavage fluid from cancer-negative cases [144], suggesting that genes become inactivated by different mechanisms at different times. Similarly, in the case of CDKN2A (p16INK4a), biallelic inactivation through LOH combined with hypermethylation was seen in 22% of ADCs, providing a mechanism for progressive CDKN2A deregulation [145]. The p16 gene encodes cell cycle proteins (inhibitor of CDK4 and CDK6) and negatively regulates cyclin D-dependent phosphorylation of the Rb gene product, thereby inhibiting cell cycle progression from G1 to S-phase by sequestration of E2F [146,147]. These observations imply that deletions at chromosomal loci 5q, 9p, 11q, and 13q are relatively early events, which suggests that inactivation of the APC, p16 (CDKN2A), INT2, and Rb genes might be functionally associated with the pathogenesis of lung ADC and also indicate that deletions of 3p, 17p, 18q, and 22q increase significantly during the course of malignant progression (Figure 3).
Epigenetic Alteration
Silencing of various tumor suppressor genes by epigenetic alteration is also an important mechanism in human carcinogenesis [148]. Epigenetic changes in one allele and LOH or another epigenetic changes of the remaining allele can also result in biallelic inactivation of tumor suppressor genes [149-152]. Aberrant DNA methylation is a typical epigenetic change that has been extensively detected in nearly all types of cancer [153-158], including lung cancer [156-158]. DNA hypermethylation mainly occurs in the CpG islands located in the promoter regions of tumor suppressor genes, effectively silencing the gene without any accompanying alterations in the DNA sequence [153]. This phenomenon is very widely observed in lung ADC, which suggests that DNA hypermethylation plays a critical role in the pathogenesis of lung ADC.Although DNA methylation is common in lung ADC, it is still unknown how frequently a specific gene is methylated at different steps during the multistep cancer progression. This is partly because AAH lesions provide little DNA due to their limited size. Nonetheless, many researchers have tried to determine the biological implication of DNA hypermethylation in the multistep tumor progression by comparing the frequencies at which loci are aberrantly methylated between noninvasive and invasive tumors. Licchesi et al. observed significant increase of hypermethylation of p16INK4a, DAPK, MGMT, RAR, RASSF1A, and hTERT genes in the histologic progression from normal to AAH, with low grade or high grade atypia and finally adenocaricnoma [80]. Tanaka et al. reported that the aberrant methylation of p16INK4a was significantly more frequent in invasive tumors (Noguchi type C) than in noninvasive tumors (Noguchi type A or B) [159]. Kubo el al. showed that the aberrant hypermethylation of p16INK4a, RASSF1A and CDH13 was significantly more frequent in invasive tumors (Noguchi type C) than in noninvasive tumors (Noguchi type A or B) using a methylation-specific PCR assay and this finding suggests that methylation of these genes play roles in the development of late-stage lung ADC, especially the acquirement of invasiveness [160].
Recently, Chung et al. showed that aberrant methylation of HOXA1, TMEFF2 and RARB occurred in noninvasive stages (AAH and AIS) and aberrant methylation of PENK, BCL2, RUNX3, DLEC1, MT1G, GRIN2B, CDH13, CCND2, and HOXA10 was more closely associated with the invasive stage than the noninvasive stage. These findings suggest that promoter CpG island hypermethylation occurs at an early stage of multistep pulmonary ADC (ADC) development and accumulates to the progression of ADC [161]. Selamat et al. performed a more comprehensive analysis for DNA methylation levels of histologically normal adjacent non-tumor lung, AAH, AIS, and invasive ADC at 15 CpG islands that are frequently affected in lung ADC using sensitive real-time PCR [162]. Loci in which DNA hypermethylation occurred in AAH (CDKN2A and PTPRN2) were different from those in AIS (2C35, EYA4, HOXA1, HOXA11, NEUROD1, NEUROD2 and TMEFF2) and invasive ADC (CDH13, CDX2, OPCML, RASSF1, SFRP1 and TWIST1) (Figure 3). This finding suggests that DNA hypermethylation of distinct loci develops at different time points during the development of lung ADC. Moreover, the fact that the number of methylated loci gradually increased from AAH to AIS and invasive ADC supports a model in which AAH and AIS are precursor lesions of a subset of lung ADCs.
Future Direction
Since the concept of multistep development and progression of lung ADC was postulated, supporting evidence has been accumulating in clinical, radiologic, pathologic and molecular studies. As mentioned earlier, however, a critical challenge that researchers are still facing in studying the molecular basis of this concept is limited availability of tissue samples. This is mainly due to the limited size of these early lesions like AAH, which are by definition smaller than 5 mm and contain few cells from their alveolar structure [17]. Another reason is related to the fact that clinicians are reluctant to perform a surgery for early indolent nodules with GGO features. A possible solution to overcome this limitation is to find an alternative way of obtaining tissue samples by AAH or AIS-derived cell lines. This interesting approach was done by Shimada and coworkers, who compared a cell line derived from an AAH lesion (PL16T) with its normal counterpart (PL16B) [163]. Although there is still concern whether these cells maintain their AAH or AIS characteristics [66], this alternative method will potentially shed light on how to study the molecular alterations occurring during the development and progression from AAH to AIS and ADC. Another way to solve the limited specimen issue is to make better use of it by high-throughput technologies such as next generation sequencing (NGS) and tissue microarray. Data generated by high-throughput experiments need to be further analyzed by bioinformatics methods.Apart from understanding the molecular basis of carcinogenesis, this effort to elucidate the concept of multistep progression can be used to develop a new biomarker specific for the different developmental stages of lung ADC. Moreover, considering that some epigenetic hits might be reversible in principle, newly-detected molecular alterations can be utilized to function as a potential therapeutic target or a great guide to chemoprevention.
Acknowledgements
This work as supported by the Barbara Isackson Lung Cancer Research Fund (DJ), The Eileen D. Ludwig Endowed Fund for Thoracic Oncology Research (DJ), Uniting Against Lung Cancer (UALC) (IJK), and Mesothelioma Applied Research Foundation (MARF) (IJK).References
- Boyle P, Levin B (2008) World Cancer Report 2008. International Agency for Research on Cancer, Lyon, France.
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, et al. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893-2917.
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, et al. (2005) Cancer statistics, 2005. CA Cancer J Clin 55: 10-30.
- Carney DN (2002) Lung cancer-time to move on from chemotherapy. N Engl J Med 346: 126-128.
- Martini N, Bains MS, Burt ME, Zakowski MF, McCormack P, et al. (1995) Incidence of local recurrence and second primary tumors in resected stage I lung cancer. J Thorac Cardiovasc Surg 109: 120-129.
- Harpole DH Jr, Herndon JE 2nd, Young WG Jr, Wolfe WG, Sabiston DC Jr (1995) Stage I non-small cell lung cancer. A multivariate analysis of treatment methods and patterns of recurrence. Cancer 76: 787-796.
- Al-Kattan K, Sepsas E, Fountain SW, Townsend ER (1997) Disease recurrence after resection for stage I lung cancer. Eur J Cardiothorac Surg 12: 380-384.
- Martin J, Ginsberg RJ, Venkatraman ES, Bains MS, Downey RJ, et al. (2002) Long-term results of combined-modality therapy in resectable non-small-cell lung cancer. J Clin Oncol 20: 1989-1995.
- Winton T, Livingston R, Johnson D, Rigas J, Johnston M, et al. (2005) Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med 352: 2589-2597.
- Jones DR, Daniel TM, Denlinger CE, Rundall BK, Smolkin ME, et al. (2006) Stage IB nonsmall cell lung cancers: are they all the same? Ann Thorac Surg 81: 1958-1962.
- Sugimura H, Nichols FC, Yang P, Allen MS, Cassivi SD, et al. (2007) Survival after recurrent nonsmall-cell lung cancer after complete pulmonary resection. Ann Thorac Surg 83: 409-417.
- Nakagawa T, Okumura N, Ohata K, Igai H, Matsuoka T, et al. (2008) Postrecurrence survival in patients with stage I non-small cell lung cancer. Eur J Cardiothorac Surg 34: 499-504.
- Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC (2004) World Health Organization classification of tumors: pathology and genetics, tumours of lung, pleura, thymus and heart. International Agency for Research on Cancer, Lyon, France.
- Curado MP, Edwards B, Shin HR, Storm H, Ferlay J, et al. (2007) Cancer Incidence in Five Continents, Vol. IX. International Agency for Research on Cancer, Lyon, France.
- Kitamura H, Kameda Y, Ito T, Hayashi H (1999) Atypical adenomatous hyperplasia of the lung. Implications for the pathogenesis of peripheral lung adenocarcinoma. Am J Clin Pathol 111: 610-622.
- Chapman AD, Kerr KM (2000) The association between atypical adenomatous hyperplasia and primary lung cancer. Br J Cancer 83: 632-636.
- Kerr KM (2001) Pulmonary preinvasive neoplasia. J Clin Pathol 54: 257-271.
- Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, et al. (2011) International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 6: 244-285.
- Westra WH (2000) Early glandular neoplasia of the lung. Respir Res 1: 163-169.
- Soda H, Nakamura Y, Nakatomi K, Tomonaga N, Yamaguchi H, et al. (2008) Stepwise progression from ground-glass opacity towards invasive adenocarcinoma: long-term follow-up of radiological findings. Lung Cancer 60: 298-301.
- Min JH, Lee HY, Lee KS, Han J, Park K, et al. (2010) Stepwise evolution from a focal pure pulmonary ground-glass opacity nodule into an invasive lung adenocarcinoma: An observation for more than 10 years. Lung Cancer 69: 123-126.
- Sone S, Takashima S, Li F, Yang Z, Honda T, et al. (1998) Mass screening for lung cancer with mobile spiral computed tomography scanner. Lancet 351: 1242-1245.
- Henschke CI, McCauley DI, Yankelevitz DF, Naidich DP, McGuinness G, et al. (1999) Early Lung Cancer Action Project: overall design and findings from baseline screening. Lancet 354: 99-105.
- Sone S, Li F, Yang ZG, Honda T, Maruyama Y, et al. (2001) Results of three-year mass screening programme for lung cancer using mobile low-dose spiral computed tomography scanner. Br J Cancer 84: 25-32.
- Sobue T, Moriyama N, Kaneko M, Kusumoto M, Kobayashi T, et al. (2002) Screening for lung cancer with low-dose helical computed tomography: anti-lung cancer association project. J Clin Oncol 20: 911-920.
- National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, et al. (2011) Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 365: 395-409.
- Park CM, Goo JM, Lee HJ, Lee CH, Chun EJ, et al. (2007) Nodular ground-glass opacity at thin-section CT: histologic correlation and evaluation of change at follow-up. Radiographics 27: 391-408.
- Godoy MC, Naidich DP (2009) Subsolid pulmonary nodules and the spectrum of peripheral adenocarcinomas of the lung: recommended interim guidelines for assessment and management. Radiology 253: 606-622.
- Nakata M, Saeki H, Takata I, Segawa Y, Mogami H, et al. (2002) Focal ground-glass opacity detected by low-dose helical CT. Chest 121: 1464-1467.
- Infante M, Lutman RF, Imparato S, Di Rocco M, Ceresoli GL, et al. (2009) Differential diagnosis and management of focal ground-glass opacities. Eur Respir J 33: 821-827.
- Jang HJ, Lee KS, Kwon OJ, Rhee CH, Shim YM, et al. (1996) Bronchioloalveolar carcinoma: focal area of ground-glass attenuation at thin-section CT as an early sign. Radiology 199: 485-488.
- Henschke CI, Yankelevitz DF, Mirtcheva R, McGuinness G, McCauley D, et al. (2002) CT screening for lung cancer: frequency and significance of part-solid and nonsolid nodules. AJR Am J Roentgenol 178: 1053-1057.
- Nakajima R, Yokose T, Kakinuma R, Nagai K, Nishiwaki Y, et al. (2002) Localized pure ground-glass opacity on high-resolution CT: histologic characteristics. J Comput Assist Tomogr 26: 323-329.
- Noguchi M, Morikawa A, Kawasaki M, Matsuno Y, Yamada T, et al. (1995) Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer 75: 2844-2852.
- Kim HY, Shim YM, Lee KS, Han J, Yi CA, et al. (2007) Persistent pulmonary nodular ground-glass opacity at thin-section CT: histopathologic comparisons. Radiology 245: 267-275.
- Travis WD, Garg K, Franklin WA, Wistuba II, Sabloff B, et al. (2005) Evolving concepts in the pathology and computed tomography imaging of lung adenocarcinoma and bronchioloalveolar carcinoma. J Clin Oncol 23: 3279-3287.
- Travis WD, Colby TV, Corrin B (1999) Histological Typing of Lung Cancer and Pleural Tumors (3rd ed). WHO International Histological Classification of Tumors, Berlin, Germany.
- Travis WD, Garg K, Franklin WA, Wistuba II, Sabloff B, et al. (2006) Bronchioloalveolar carcinoma and lung adenocarcinoma: the clinical importance and research relevance of the 2004 World Health Organization pathologic criteria. J Thorac Oncol 1: S13-S19.
- Watanabe S, Watanabe T, Arai K, Kasai T, Haratake J, et al. (2002) Results of wedge resection for focal bronchioloalveolar carcinoma showing pure ground-glass attenuation on computed tomography. Ann Thorac Surg 73: 1071-1075.
- Sakurai H, Dobashi Y, Mizutani E, Matsubara H, Suzuki S, et al. (2004) Bronchioloalveolar carcinoma of the lung 3 centimeters or less in diameter: a prognostic assessment. Ann Thorac Surg 78: 1728-1733.
- Vazquez M, Carter D, Brambilla E, Gazdar A, Noguchi M, et al. (2009) Solitary and multiple resected adenocarcinomas after CT screening for lung cancer: histopathologic features and their prognostic implications. Lung Cancer 64: 148-154.
- Yamato Y, Tsuchida M, Watanabe T, Aoki T, Koizumi N, et al. (2001) Early results of a prospective study of limited resection for bronchioloalveolar adenocarcinoma of the lung. Ann Thorac Surg 71: 971-974.
- Yamada S, Kohno T (2004) Video-assisted thoracic surgery for pure ground- glass opacities 2 cm or less in diameter. Ann Thorac Surg 77: 1911-1915.
- Yoshida J, Nagai K, Yokose T, Nishimura M, Kakinuma R, et al. (2005) Limited resection trial for pulmonary ground-glass opacity nodules: fifty-case experience. J Thorac Cardiovasc Surg 129: 991-996.
- Koike T, Togashi K, Shirato T, Sato S, Hirahara H, et al. (2009) Limited resection for noninvasive bronchioloalveolar carcinoma diagnosed by intraoperative pathologic examination. Ann Thorac Surg 88: 1106-1111.
- Takashima S, Maruyama Y, Hasegawa M, Yamanda T, Honda T, et al. (2003) CT findings and progression of small peripheral lung neoplasms having a replacement growth pattern. AJR Am J Roentgenol 180: 817-826.
- Kakinuma R, Ohmatsu H, Kaneko M, Kusumoto M, Yoshida J, et al. (2004) Progression of focal pure ground-glass opacity detected by low-dose helical computed tomography screening for lung cancer. J Comput Assist Tomogr 28: 17-23.
- Ko JP (2005) Lung nodule detection and characterization with multi-slice CT. J Thorac Imaging 20: 196-209.
- Yang ZG, Sone S, Takashima S, Li F, Honda T, et al. (2001) High-resolution CT analysis of small peripheral lung adenocarcinomas revealed on screening helical CT. AJR Am J Roentgenol 176: 1399-1407.
- Asamura H, Suzuki K, Watanabe S, Matsuno Y, Maeshima A, et al. (2003) A clinicopathological study of resected sub-centimeter lung cancers: a favorable prognosis for ground glass opacity lesions. Ann Thorac Surg 76: 1016-1022.
- Kondo T, Yamada K, Noda K, Nakayama H, Kameda Y (2002) Radiologic-prognostic correlation in patients with small pulmonary adenocarcinomas. Lung Cancer 36: 49-57.
- Takashima S, Maruyama Y, Hasegawa M, Yamanda T, Honda T et al. (2002) Prognostic significance of high-resolution CT findings in small peripheral adenocarcinoma of the lung: a retrospective study on 64 patients. Lung Cancer 36: 289-295.
- Nagao M, Murase K, Yasuhara Y, Ikezoe J, Eguchi K, et al. (2002) Measurement of localized ground-glass attenuation on thin-section computed tomography images: correlation with the progression of bronchioloalveolar carcinoma of the lung. Invest Radiol 37: 692-697.
- Kodama K, Higashiyama M, Yokouchi H, Takami K, Kuriyama K, et al. (2001) Prognostic value of ground-glass opacity found in small lung adenocarcinoma on high-resolution CT scanning. Lung Cancer 33: 17-25.
- Kodama K, Higashiyama M, Yokouchi H, Takami K, Kuriyama K, et al. (2002) Natural history of pure ground-glass opacity after long-term follow-up of more than 2 years. Ann Thorac Surg 73: 386-392.
- Reich JM (2008) A critical appraisal of overdiagnosis: estimates of its magnitude and implications for lung cancer screening. Thorax 63: 377-383.
- Nakata M, Sawada S, Saeke H, Takashima S, Mogami H, et al. (2003) Prospective study of thoracoscopic limited resection for ground-glass opacity selected by computed tomography. Ann Thorac Surg 75: 1601-1606.
- Asamura H, Suzuki K, Watanabe S, Matsuno Y, Maeshima A, et al. (2003) A clinicopathological study of resected subcentimeter lung cancers: a favorable prognosis for ground glass opacity lesions. Ann Thorac Surg 76: 1016-1022.
- Watanabe S, Watanabe T, Arai K, Kasai T, Haratake J, et al. (2002) Results of wedge resection for focal bronchioloalveolar carcinoma showing pure ground-glass attenuation on computed tomography. Ann Thorac Surg 73: 1071-1075.
- Borczuk AC, Qian F, Kazeros A, Eleazar J, Assaad A, et al. (2009) Invasive size is an independent predictor of survival in pulmonary adenocarcinoma. Am J Surg Pathol 33: 462-469.
- Yim J, Zhu LC, Chiriboga L, Watson HN, Goldberg JD, et al. (2007) Histologic features are important prognostic indicators in early stages lung adenocarcinomas. Mod Pathol 20: 233-241.
- Goldstein NS, Mani A, Chmielewski G, Welsh R, Pursel S (1999) Prognostic factors in T1 NO MO adenocarcinomas and bronchioloalveolar carcinomas of the lung. Am J Clin Pathol 112: 391-402.
- Riquet M, Foucault C, Berna P, Assouad J, Dujon A, et al. (2006) Prognostic value of histology in resected lung cancer with emphasis on the relevance of the adenocarcinoma subtyping. Ann Thorac Surg 81: 1988-1995.
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, et al. (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319:525-532.
- Arnold CN, Goel A, Blum HE, Boland CR (2005) Molecular Pathogenesis of Colorectal Cancer. Cancer 15: 2035-2047.
- Kerr KM, Galler JS, Hagen JA, Laird PW, Laird-Offringa IA (2007) The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 23: 5-30.
- Vogelstein B, Kinzler KW (2004) Cancer genes and the pathways they control. Nat Med 10: 789-799.
- Yatabe Y, Borczuk AC, Powell CA (2011) Do all lung adenocarcinomas follow a stepwise progression? Lung cancer 74: 7-11.
- Mori M, Rao SK, Popper HH, Cagle PT, Fraire AE (2001) Atypical adenomatous hyperplasia of the lung: a probable forerunner in the development of adenocarcinoma of the lung. Mod Pathol 14: 72-84.
- Soh J, Toyooka S, Ichihara S, Asano H, Kobayashi N, et al. (2008) Sequential molecular changes during multistage pathogenesis of small peripheral adenocarcinomas of the lung. J Thorac Oncol 3: 340-347.
- Lantuejoul S, Salameire D, Salon C, Brambilla E (2009) Pulmonary preneoplasia-sequential molecular carcinogenetic events. Histopathology 54: 43-54.
- Nakayama H, Noguchi M, Tsuchiya R, Kodama T, Shimosato Y (1990) Clonal growth of atypical adenomatous hyperplasia of the lung: cytofluorometric analysis of nuclear DNA content. Mod Pathol 3: 314-320.
- Niho S, Yokose T, Suzuki K, Kodama T, Nishiwaki Y, et al. (1999) Monoclonality of atypical adenomatous hyperplasia of the lung. Am J Pathol 154: 249-254.
- Sakamoto H, Shimizu J, Horio Y, Ueda R, Takahashi T, et al. (2007) Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasion to invasive adenocarcinoma. J Pathol 212: 287-294.
- Westra WH, Baas IO, Hruban RH, Askin FB, Wilson K, et al. (1996) K-ras oncogene activation in atypical alveolar hyperplasias of the human lung. Cancer Res 56: 2224-2228.
- Kohno T, Kunitoh H, Suzuki K, Yamamoto S, Kuchiba A, et al. (2008) Association of KRAS polymorphisms with risk for lung adenocarcinoma accompanied by atypical adenomatous hyperplasias. Carcinogenesis 29: 957-963.
- Yoshida Y, Shibata T, Kokubu A, Tsuta K, Matsuno Y, et al. (2005) Mutations of the epidermal growth factor receptor gene in atypical adenomatous hyperplasia and bronchioloalveolar carcinoma of the lung. Lung Cancer 50: 1-8.
- Kitamura H, Kameda Y, Nakamura N, Inayama Y, Nakatani Y, et al. (1996) Atypical adenomatous hyperplasia and bronchoalveolar lung carcinoma. Analysis by morphometry and the expressions of p53 and carcinoembryonic antigen. Am J Surg Pathol 20: 553-562.
- Takamochi K, Ogura T, Suzuki K, Kawasaki H, Kurashima Y, et al. (2001) Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol 159: 1941-1948.
- Licchesi JD, Westra WH, Hooker CM, Herman JG (2008) Promoter hypermethylation of hallmark cancer genes in atypical adenomatous hyperplasia of the lung. Clin Cancer Res 14: 2570-2578.
- Nakanishi K, Kawai T, Kumaki F, Hirot S, Mukai M, et al. (2002) Expression of human telomerase RNA component and telomerase reverse transcriptase mRNA in atypical adenomatous hyperplasia of the lung. Hum Pathol 33: 697-702.
- Seki N, Takasu T, Mandai K, Nakata M, Saeki H, et al. (2002) Expression of eukaryotic initiation factor 4E in atypical adenomatous hyperplasia and adenocarcinoma of the human peripheral lung. Clin Cancer Res 8: 3046-3053.
- Licchesi JD, Westra WH, Hooker CM, Machida EO, Baylin SB, et al. (2008) Epigenetic alteration of Wnt pathway antagonists in progressive glandular neoplasia of the lung. Carcinogenesis 29: 895-904.
- Kerr KM, MacKenzie SJ, Ramasami S, Murray GI, Fyfe N, et al. (2004) Expression of Fhit, cell adhesion molecules and matrix metalloproteinases in atypical adenomatous hyperplasia and pulmonary adenocarcinoma. J Pathol 203: 638-644.
- Westra WH, Slebos RJ, Offerhaus GJ, Goodman SN, Evers SG, et al. (1993) K-ras oncogene activation in lung adenocarcinomas from former smokers. Evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. Cancer 72: 432-438.
- Morandi L, Asioli S, Cavazza A, Pession A, Damiani S (2007) Genetic relationship among atypical adenomatous hyperplasia, bronchioloalveolar carcinoma and adenocarcinoma of the lung. Lung Cancer 56: 35-42.
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, et al. (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243-3248.
- Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, et al. (2006) Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20: 1496-1510.
- Minna JD, Roth JA, Gazdar AF (2002) Focus on lung cancer. Cancer Cell 1: 49-52.
- Yatabe Y, Kosaka T, Takahashi T, Mitsudomi T (2005) EGFR mutation is specific for terminal respiratory unit type adenocarcinoma. Am J Surg Pathol 29: 633-639.
- Haneda H, Sasaki H, Shimizu S, Endo K, Suzuki E, et al. (2006) Epidermal growth factor receptor gene mutation defines distinct subsets among small adenocarcinomas of the lung. Lung Cancer 52: 47-52.
- Sakuma Y, Matsukuma S, Yoshihara M, Nakamura Y, Noda K, et al. (2007) Distinctive evaluation of nonmucinous and mucinous subtypes of bronchioloalveolar carcinomas in EGFR and K-ras gene-mutation analyses for Japanese lung adenocarcinomas: confirmation of the correlations with histologic subtypes and gene mutations. Am J Clin Pathol 128: 100-108.
- Cooper CA, Carby FA, Bubb VJ, Lamb D, Kerr KM, et al. (1997) The pattern of K-ras mutation in pulmonary adenocarcinoma defines a new pathway of tumour development in the human lung. J Pathol 181: 401-404.
- Ji H, Li D, Chen L, Shimamura T, Kobayashi S, et al. (2006) The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell 9: 485-495.
- Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, et al. (2003) Epidermal growth factor receptor: mechanisms of activation and signaling. Exp Cell Res 284: 31-53.
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, et al. (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304: 1497-1500.
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, et al. (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350: 2129-2139.
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, et al. (2004) EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 101: 13306-13311.
- Huang SF, Liu HP, Li LH, Ku YC, Fu YN, et al. (2004) High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res 10: 8195-8203.
- Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, et al. (2004) Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res 64: 8919-8923.
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, et al. (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97: 339-346.
- Choi H, Kratz J, Pham P, Lee S, Ray R, et al. (2012) Development of a rapid and practical mutation screening assay for human lung adenocarcinoma. Int J Oncol 40: 1900-1906.
- Kozuki T, Hisamoto A, Tabata M, Takigawa N, Kiura K, et al. (2007) Mutation of the epidermal growth factor receptor gene in the development of adenocarcinoma of the lung. Lung Cancer 58: 30-35.
- Matsumoto S, Iwakawa R, Kohno T, Suzuki K, Matsuno Y, et al. (2006) Frequent EGFR mutations in noninvasive bronchioloalveolar carcinoma. Int J Cancer 118: 2498-2504.
- Tang X, Shigematsu H, Bekele BN, Roth JA, Minna JD, et al. (2005) EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 65: 7568-7572.
- Yoo SB, Chung JH, Lee HJ, Lee CT, Jheon S, et al. (2010) Epidermal growth factor receptor mutation and p53 overexpression during the multistage progression of small adenocarcinoma of the lung. J Thorac Oncol 5: 964-969.
- Nomura M, Shigematsu H, Li L, Suzuki M, Takahashi T, et al. (2007) Polymorphisms, mutations, and amplification of the EGFR gene in non-small cell lung cancers. PLoS Med 4: e125.
- Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, et al. (2005) Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res 65: 1642-1646.
- Kerr KM, Carey FA, King G, Lamb D (1994) Atypical adenomatous hyperplasia: relationship with pulmonary adenocarcinoma, p53, and c-erB-2 expression. J Pathol 174: 249-56.
- Tateishi M, Ishida T, Mitsudomi T, Kaneko S, Sugimachi K (1991) Prognostic value of c-erbB-2 protein expression in human lung adenocarcinoma and squamous cell carcinoma. Eur J Cancer 27: 1372-1375.
- Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR (1982) Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci U S A 79: 4848-4852.
- McCormick F (1989) ras GTPase activating protein: signal transmitter and signal terminator. Cell 56: 5-8.
- Shih C, Weinberg RA (1982) Isolation of transforming sequence from a human bladder carcinoma cell line. Cell 29: 161-169.
- Cooper GM (1982) Cellular transforming genes. Science 217: 801-806.
- Pulciani S, Santos E, Lauver AV, Long LK, Aaronson SA, et al. (1982) Oncogenes in solid human tumors. Nature 300: 539-542.
- Keohavong P, DeMichele MA, Melacrinos AC, Landreneau RJ, Weyant RJ, et al. (1996) Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res 2: 411-418.
- Rodenhuis S, Slebos RJ (1992) Clinical significance of ras oncogene activation in human lung cancer. Cancer Res 52: 2665s-2669s.
- Slebos RJ, Hruban RH, Dalesio O, Mooi WJ, Offerhaus GJ, et al. (1991) Relationship between K-ras oncogene activation and smoking in adenocarcinoma of the human lung. J Natl Cancer Inst 83: 1024-1027.
- Cho JY, Kim JH, Lee YH, Chung KY, Kim SK, et al. (1997) Correlation between K-ras gene mutation and prognosis of patients with nonsmall cell lung carcinoma. Cancer 79: 462-467.
- Fukuyama Y, Mitsudomi T, Sugio K, Ishida T, Akazawa K, et al. (1997) K-ras and p53 mutations are an independent unfavourable prognostic indicator in patients with non-small-cell lung cancer. Br J Cancer 75:1125-1130.
- Toyooka S, Tokumo M, Shigematsu H, Matsuo K, Asano H, et al. (2006) Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res 66: 1371-1375.
- Marchetti A, Buttitta F, Pellegrini S, Chella A, Bertacca G, et al. (1996) Bronchioloalveolar lung carcinomas: K-ras mutations are constant events in the mucinous subtype. J Pathol 179: 254-259.
- Tam IY, Chung LP, Suen WS, Wang E, Wong MC, et al. (2006) Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res 12: 1647-1653.
- Han SW, Kim TY, Jeon YK, Hwang PG, Im SA, et al. (2006) Optimization of patient selection for gefitinib in non-small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K-ras mutation, and Akt phosphorylation. Clin Cancer Res 12: 2538-2544.
- Mills NE, Fishman CL, Rom WN, Dubin N, Jacobson DR (1995) Increased prevalence of K-ras oncogene mutations in lung adenocarcinomas. Cancer Res 55: 1444-1447.
- Li ZH, Zheng J, Weiss LM, Shibata D (1994) c-K-ras and p53 mutations occur very early in adenocarcinoma of the lung. Am J Pathol 144: 303-309.
- Kumar R, Sukumar S, Barbacid M (1990) Activation of ras oncogenes preceding the onset of neoplasia. Science 248: 1101-1104.
- Ohshima S, Shimizu Y, Takahama M (1994) Detection of c-Ki-ras gene mutation in paraffin sections of adenocarcinoma and atypical bronchioloalveolar cell hyperplasia of human lung. Virchows Arch 424: 129-134.
- Sugio K, Kishimoto Y, Virmani AK, Hung JY, Gazdar AF (1994) K-ras mutations are a relatively late event in the pathogenesis of lung carcinomas. Cancer Res 54: 5811-5815.
- Sakuma Y, Matsukuma S, Yoshihara M, Nakamura Y, Nakayama H, et al. (2007) Epidermal growth factor receptor gene mutations in atypical adenomatous hyperplasia of the lung. Mod Pathol 20: 967-973.
- Harris CC, Hollstein M (1993) Clinical implications of the p53 tumor-suppressor gene. N Engl J Med 329: 1318-1327.
- Wistuba II, Behrens C, Virmani AK, Mele G, Milchgrub S, et al. (2000) High resolution chromosome 3p allelotyping of human lung cancer and preneoplastic/preinvasive bronchial epithelium reveals multiple, discontinuous sites of 3p allele loss and three regions of frequent breakpoints. Cancer Res 60: 1949-1960.
- Zabarovsky ER, Lerman MI, Minna JD (2002) Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 21: 6915-6935.
- Grepmeier U, Dietmaier W, Merk J, Wild PJ, Obermann EC, et al. (2005) Deletions at chromosome 2q and 12p are early and frequent molecular alterations in bronchial epithelium and NSCLC of long-term smokers. Int J Oncol 27: 481-488.
- Sanchez-Cespedes M, Ahrendt SA, Piantadosi S, Rosell R, Monzo M, et al. (2001) Chromosomal alterations in lung adenocarcinoma from smokers and nonsmokers. Cancer Res 61: 1309-1313.
- Wistuba II, Mao L, Gazdar AF (2002) Smoking molecular damage in bronchial epithelium. Oncogene 21: 7298-7306.
- Mao L, Lee JS, Kurie JM, Fan YH, Lippman SM, et al. (1997) Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst 89: 857-862.
- Wistuba II, Lam S, Behrens C, Virmani AK, Fong KM, et al. (1997) Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst 89: 1366-1373.
- Aoyagi Y, Yokose T, Minami Y, Ochiai A, Iijima T, et al. (2001) Accumulation of losses of heterozygosity and multistep carcinogenesis in pulmonary adenocarcinoma. Cancer Res 61: 7950-7954.
- Kitaguchi S, Takeshima Y, Nishisaka T, Inai K (1998) Proliferative activity, p53 expression and loss of heterozygosity on 3p, 9p and 17p in atypical adenomatous hyperplasia of the lung. Hiroshima J Med Sci 47: 17-25.
- Kohno H, Hiroshima K, Toyozaki T, Fujisawa T, Ohwada H (1999) p53 mutation and allelic loss of chromosome 3p, 9p of preneoplastic lesions in patients with nonsmall cell lung carcinoma. Cancer 85: 341-347.
- Yamasaki M, Takeshima Y, Fujii S, Kitaguchi S, Matsuura M, et al. (2000) Correlation between genetic alterations and histopathological subtypes in bronchiolo-alveolar carcinoma and atypical adenomatous hyperplasia of the lung. Pathol Int 50: 778-785.
- Takamochi K, Ogura T, Suzuki K, Kawasaki H, Kurashima Y, et al. (2001) Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol 159: 1941-1948.
- de Fraipont F, Moro-Sibilot D, Michelland S, Brambilla E, Brambilla C, et al. (2005) Promoter methylation of genes in bronchial lavages: a marker for early diagnosis of primary and relapsing non-small cell lung cancer? Lung Cancer 50: 199-209.
- Awaya H, Takeshima Y, Amatya VJ, Furonaka O, Tagawa K, et al. (2004) Inactivation of the p16 gene by hypermethylation and loss of heterozygosity in adenocarcinoma of the lung. Pathol Int 54: 486-489.
- Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, et al. (1994) Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature 368: 753-756.
- Kamb A, Gruis NA, Weaverfeldhaus J, Liu Q, Harshman K, et al. (1994) A cell-cycle regulator potentially involved in genesis of many tumor types. Science 264: 436-440.
- Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP (1998) Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res 72: 141-196.
- Garinis GA, Patrinos GP, Spanakis NE, Menounos PG (2002) DNA hypermethylation: when tumour suppressor genes go silent. Hum Genet 111: 115-127.
- Tucker T, Friedman JM (2002) Pathogenesis of hereditary tumors: beyond the ‘two-hit’ hypothesis. Clin Genet 62: 345-357.
- Jone PA, Laird PW (1999) Cancer-epigenetics comes of age. Nature Genet 21: 163-167.
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247-257.
- Jones PA, Baylin SB (2002) The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415-428.
- Costello JC, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, et al. (2000) Aberrant CpG-island methylation has non-random and tumor-type-specific patterns. Nature Genet 25: 132-138.
- Esteller M, Corn PG, Baylin SB, Herman JG (2001) A gene hypermethylation profile of human cancer. Cancer Res 61: 3225-3229.
- Tsou JA, Hagen JA, Carpenter CL, Laird-Offringa IA (2002) DNA methylation analysis: a powerful new tool for lung cancer diagnosis. Oncogene 21: 5450-5461.
- Belinsky SA (2004) Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer 4: 707-717.
- Digel W, Lubbert M (2005) DNA methylation disturbances as novel therapeutic target in lung cancer: preclinical and clinical results. Crit Rev Oncol Hematol 55: 1-11.
- Tanaka R, Wang D, Morishita Y, Inadome Y, Minami Y, et al. (2005) Loss of function of p16 gene and prognosis of pulmonary adenocarcinoma. Cancer 103: 608-615.
- Kubo T, Yamamoto H, Ichimura K, Jida M, Hayashi T, et al. (2009) DNA methylation in small lung adenocarcinoma with bronchioloalveolar carcinoma components. Lung Cancer 65: 328-332.
- Chung JH, Lee HJ, Kim BH, Cho NY, Kang GH (2011) DNA methylation profile during multistage progression of pulmonary adenocarcinomas. Virchows Arch 459: 201-211.
- Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, et al. (2011) DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS One 6: e21443.
- Shimada A, Kano J, Ishiyama T, Okubo C, Iijima T, et al. (2005) Establishment of an immortalized cell line from a precancerous lesion of lung adenocarcinoma, and genes highly expressed in the early stages of lung adenocarcinoma development. Cancer Sci 96: 668-675.