
Journal of Urology & Nephrology
Download PDF
Hsp90 N-domain inhibitors

The new-generation resorcinol derivative ganetespib is in a Phase II trial of mCRPC (NCT01270880, www.clinicaltrials.gov) inpatients that have previously been treated with docetaxel. The primary outcome measure will be progression-free survival (PFS), with secondary outcomes of PSA decrease, overall survival, association of PFS and PSA, and elucidation of markers of drug response. Ganetespib is in a Phase III trial (NCT01798485,www.clinicaltrials.gov) in combination with docetaxel for the treatment of advanced non-small cell lung cancer and hasdemonstrated an improved safety profile in patient populations as compared to first generation inhibitors [36]. Ganetespib is also in clinical trials for the treatment of cancers of the liver, breast, malignant peripheral nerve sheath, ovary, and fallopian tube as wellas myelodysplastic syndrome.
Tumors that are addicted to Hsp90 clients such as HER2 in breast cancer and EML4-ALK in non-small cell lung cancer (NSCLC) show clinical responses to Hsp90 inhibitors [14]. Initial preclinical observations suggested that CRPC may respond favorably to Hsp90 inhibitor therapy because AR activity is one of the major factors for disease progression. However Hsp90 inhibition as a single agent, though potent in the laboratory, has shown numerous clinical limitations in patients with CRPC. Therefore these drugs are likely to be most effective when used as part of combination treatments.
Research Article
*Address for Correspondence: Mehdi Mollapour, Department of Urology, SUNY Upstate Medical University 750 East Adams St., Syracuse, NY, 13210, USA, E-mail: mollapom@upstate.edueptor (AR) [4]
Citation: Woodford MR, Madala A, Schulman J, Nsouli T, Mollapour M. Efficacy of the Hsp90 Inhibitors in Prostate Cancer Therapy. J Urol Nephrol. 2014;1(1): 9.eptor (AR) [4].
Copyright © 2014 Woodford MR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.eptor (AR) [4].
Journal of Urology & Nephrology | ISSN 2380-0585 | Volume: 1, Issue: 1eptor (AR) [4].
Submission: 21 February 2014 | Accepted: 21 March 2014 | Published: 03 April 2014eptor (AR) [4].
Reviewed & Approved by: Dr. Sanjeev Shukla, Department of Urology, Case Western Reserve University, USA
Keywords
Abbreviation: AR: Androgen Receptor; ARE: Androgen Response Element; CRPC: Castrate-Resistant Prostate Cancer; DHT: Dihydrotestosterone; Hsp90: Central Molecular Chaperone Involved in AR Stabilization and Activity; NF- κb: Nuclear Factor Kappa-Light-Chain-Enhancer of activated B Cells, Transcription Factor Involved in Cytokine Expression; TNF: Tumor Necrosis Factor; TRAIL: TNF-Related Apoptosis-Inducing Ligand, Induces Apoptotic Cell Death; TRAMP: Transgenic Adenocarcinoma of the Mouse Prostate; PFS: Progression-Free Survival
Hsp90 is an essential molecular chaperone in eukaryotes and it is critical for folding and/or degradation of approximately 200 proteins referred to as “clients” (www.picard.ch/downloads/downloads.htm)[15]. A majority of these clients are necessary for normal cellular function [16], however cancer cells use the Hsp90 chaperone machinery to protect numerous mutated or overexpressed oncoproteins from misfolding and degradation [17]. Therefore, Hsp90 has proven to be an attractive target in cancer therapy.
Efficacy of the Hsp90 Inhibitors in Prostate Cancer Therapy
Mark R. Woodford1, Alosh Madala1, Jacqualyn Schulman1,2, Tamara Nsouli1 and Mehdi Mollapour1,2,3*
- 1Department of Urology, SUNY Upstate Medical University 750 East Adams St., Syracuse, NY, 13210, USA
- 2Department of Biochemistry and Molecular Biology, SUNY Upstate Medical University 750 East Adams St., Syracuse, NY, 13210, USA
- 3Cancer Research Institute, SUNY Upstate Medical University 750 East Adams St., Syracuse, NY, 13210, USA
*Address for Correspondence: Mehdi Mollapour, Department of Urology, SUNY Upstate Medical University 750 East Adams St., Syracuse, NY, 13210, USA, E-mail: mollapom@upstate.edueptor (AR) [4]
Citation: Woodford MR, Madala A, Schulman J, Nsouli T, Mollapour M. Efficacy of the Hsp90 Inhibitors in Prostate Cancer Therapy. J Urol Nephrol. 2014;1(1): 9.eptor (AR) [4].
Copyright © 2014 Woodford MR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.eptor (AR) [4].
Journal of Urology & Nephrology | ISSN 2380-0585 | Volume: 1, Issue: 1eptor (AR) [4].
Submission: 21 February 2014 | Accepted: 21 March 2014 | Published: 03 April 2014eptor (AR) [4].
Reviewed & Approved by: Dr. Sanjeev Shukla, Department of Urology, Case Western Reserve University, USA
Abstract
Prostate cancer is a disease of aging and the second leading cause of death in men in the United States. A distinctive characteristic of prostate tumors is their dependence on androgen for development, growth and survival. The molecular chaperone Heat Shock Protein-90 (Hsp90) is involved in the stability of a number of protein targets including androgen receptor (AR). Preclinical data demonstrates Hsp90 inhibition to be an effective therapy for inhibiting prostate cancer cells in culture as well as in animal xenograft models. Two newgeneration Hsp90 inhibitors, ganetespib and AT13387, are currently being evaluated as single or combinational therapies in the clinic for castrate resistant prostate cancer (CRPC). While clinical signs of antitumor activity have been observed with new-generation HSP90 inhibitors, these drugs are likely to be most effective when used as part of combination treatments. This review highlights how inhibition of Hsp90 combined with specific secondary targets including Wee1, Hsp27, ERK and mTOR displays enhanced inhibitory effects in pre-clinical models. One clinical benefit of co-inhibition is the targeting of multiple signaling pathways. In addition, the variety of proposed combination therapies allows for latitude in selecting a secondary target, potentially tailoring therapies to specific patient populations.Keywords
Heat shock protein 90; Molecular chaperones; Castrateresistantprostate cancer; Anti-cancer drugs
Abbreviation: AR: Androgen Receptor; ARE: Androgen Response Element; CRPC: Castrate-Resistant Prostate Cancer; DHT: Dihydrotestosterone; Hsp90: Central Molecular Chaperone Involved in AR Stabilization and Activity; NF- κb: Nuclear Factor Kappa-Light-Chain-Enhancer of activated B Cells, Transcription Factor Involved in Cytokine Expression; TNF: Tumor Necrosis Factor; TRAIL: TNF-Related Apoptosis-Inducing Ligand, Induces Apoptotic Cell Death; TRAMP: Transgenic Adenocarcinoma of the Mouse Prostate; PFS: Progression-Free Survival
Introduction
Prostate cancer is one of the most common solid neoplasms and the second leading cause of death due to cancer in men in the United States [1-3]. The normal prostate as well as early-stage prostate cancer relies on androgens (eg. testosterone and its metabolite 5-dihydrotestosterone (DHT)) and their binding to androgen receptor (AR) [4].Treatment options for clinically localized prostate cancer include active surveillance (watchful waiting), interstitial prostate brachytherapy, external beam radiotherapy, radical prostatectomy and primary hormone therapy (androgen deprivation therapy, ADT). The goal of ADT is to lower serum testosterone to castrate levels. This can be accomplished either by surgical orchiectomy (castration) or medical orchiectomy by using a gonadotropin releasing hormone (GnRH) agonist or a GnRH antagonist. In some cases, anti-androgens can be combined with a GnRH agonist to provide a combined androgen blockade [5].
However, treatment is usually marked by progression to castrationresistant prostate cancer (CRPC) over a period of 18 months, with a median survival of 1-2 years. Prostate cancer deaths are usually the result of metastatic castration-resistant prostate cancer (mCRPC).
The exact mechanism of transition from castration-sensitive prostate cancer to castration-resistant prostate cancer is poorly understood, however AR remains active and continues to drive prostate cancer progression despite castrate levels of androgens.
This understanding has led to the development of multiple novel agents that are aimed at further decreasing androgen production or blocking AR function.
The treatment of men with mCRPC has changed dramatically over the past 10 years. Prior to 2004, once patients failed primary ADT they were provided palliative care. Berthold et al. and Petrylak et al. [6,7] demonstrated that docetaxel, an FDA-approved antimitotic chemotherapeutic agent, improved survival in these patients [6,7]. Since the approval of docetaxel, other agents have been shown to have a survival benefit and have been FDA approved on the basis of randomized controlled trials. These include Radium 223 [8], a radiopharmaceutical that targets bone metastases, enzalutamide (MDV3100) [9] and abiraterone [10], which specifically affect the androgen axis, sipuleucel-T [11] that stimulates the immune system, and cabazitaxel [12], a chemotherapeutic agent. However, resistance to these new agents, linked to continued hormone-driven oncogenesis has been reported [13]. Thus the incomplete efficacy of ADT highlights an urgent need for alternative treatment strategies.
In this regard, targeting the molecular chaperone heat shock protein 90 (Hsp90) has emerged as a potential avenue for therapeutic intervention [14].
Structural and Functional Architecture of Hsp90
Hsp90 exists as a homodimer, with each monomer made up of three domains: an N-terminal domain with an ATP-, drug- and cochaperone binding site, a middle domain for client and co-chaperone binding, and a C-terminal domain containing a dimerization region, and further site for client, drug and co-chaperone binding [16,18,19], Hsp90’s ATP-driven chaperoning activity, associated conformational modulations, and co-chaperone interactions are collectively known as the chaperone cycle [20] (Figure 1). This chaperone cycle follows a carefully orchestrated pattern of open and closed conformations that alter the stability of client proteins [21]. Many Hsp90 inhibitors target this process, mainly the open conformation, and competitively inhibit ATP binding, therefore destabilizing and degrading client proteins [22,23]. This has made Hsp90 an attractive target for therapeutic intervention in cancers. Initial preclinical observations suggested that CRPC might respond favorably to Hsp90 inhibitors because the activity of several Hsp90 clients, including AR, remains crucial for disease progression.
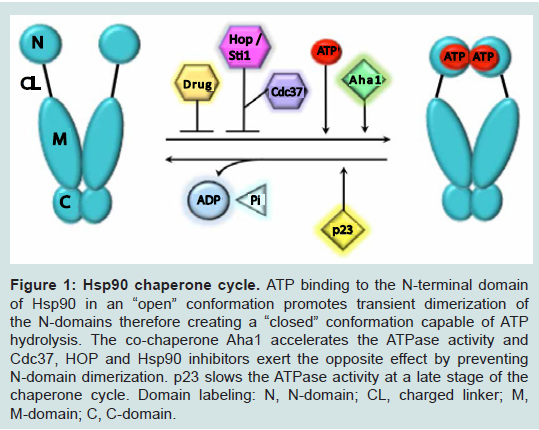
Figure 1: Hsp90 chaperone cycle.
ATP binding to the N-terminal domain of Hsp90 in an “open” conformation promotes transient dimerization of the N-domains therefore creating a “closed” conformation capable of ATP hydrolysis. The co-chaperone Aha1 accelerates the ATPase activity and Cdc37, HOP and Hsp90 inhibitors exert the opposite effect by preventingN-domain dimerization. p23 slows the ATPase activity at a late stage of the chaperone cycle. Domain labeling: N, N-domain; CL, charged linker; M,M-domain; C, C-domain.
Small Molecule Inhibitors of Hsp90
Many Hsp90 inhibitors function by mimicking the conformation adopted by ATP upon binding the N-domain of Hsp90 [23,24]. This prevents ATP binding and hydrolysis, generally leading to proteasomal degradation of client proteins. Early Hsp90 inhibitors such as RD (radicicol, a macrocyclic antifungal antibiotic) and GA (geldanamycin, a benzaquinoid ansamycin antibiotic), though potent, possessed undesirable characteristics, namely poor solubility, instability, and high toxicity at therapeutic doses [14]. This led to the derivation and development of synthetic new-generation compounds, many of which have participated in clinical trials for various cancers. Two of these compounds are in active clinical trials combating prostate cancer (www.clinicaltrials.gov) and will be discussed later.There are other inhibitors that do not target and bind to the Hsp90 N-domain. For example sulphoxythiocarbamates modify cysteine residues of the M-domain causing a conformational change that inhibits client binding, but not ATP hydrolysis [25]. Novobiocin andits derivatives such as KU32, KU135 and KU174, as well as gedunin and celastrol bind to the C-domain inhibiting interaction with cochaperones and clients [26-28]. For the purposes of this review, we will focus only on inhibitors that have shown efficacy against prostate cancer. For a more in-depth review of Hsp90 inhibitors refer to [29,30].
Pre-Clinical Evaluation of Hsp90 Inhibitors as a Monotherapy
Tanespimycin (17-N-Allylamino-17-demethoxygeldanamycin, 17-AAG) is a first-generation Hsp90 inhibitor that promotes AR degradation in prostate cancer cell culture and in mouse xenograft models [31]. Furthermore, 17-AAG-treated LuCaP35 xenograft tumors were delayed in their transition to CRPC as compared to untreated due to disruption of the mechanism by which AR translocates to the nucleus [32,33].
NVP-AUY922 is a new-generation synthetic small molecule inhibitor of Hsp90 that has shown clinical efficacy in multiple tumor lines and PC3-LN3 tumor xenografts in mice [34]. NVP-AUY922 and NVP-HSP990, another synthetic small molecule N-terminal Hsp90 inhibitor, showed similar activity to 17-AAG with respect to degradation of client proteins and Hsp70 induction in ex vivo cultured primary prostate tumors. The two synthetic compounds did show greater efficacy than 17-AAG in causing G2-M cell cycle arrest, reducing cell proliferation and inducing apoptotic cell death [35]. This study also showed that Hsp70 induction via Hsp90 inhibition is not necessarily correlated to clinical outcome despite Hsp70’s protective properties [36].
Ganetespib, a new-generation Hsp90 inhibitor, is currently undergoing evaluation in 14 clinical trials, including two related phase IIb/III and phase III studies in advanced NSCLC adenocarcinoma in combination with docetaxel (NCT01348126 and NCT01798485) [36].
Ganetespib has been shown to induce apoptosis in both androgen sensitive and insensitive cell lines (LNCaP, VCaP, Du145, and PC3) [37]. Of note, GA and ganetespib demonstrate differential effects in these prostate cancer cell lines [37], perhaps underscoring the need for these more potent Hsp90 inhibitors in the clinic. PC3 and 22Rv1 xenografts in mice experienced growth delay with weekly treatment of ganetespib and 22Rv1 cell culture, enriched for expression of the AR ligand-incompetent transcript variant V7, also displayed sensitivity to ganetespib [37]. This sensitivity occurred despite the lack of ganetespib-mediated V7 degradation, as these variants do not require Hsp90 complex formation for ligand binding [38]. Though full-length AR is a known Hsp90 client (Figure 2), data have been presented demonstrating enrichment of multiple non-Hsp90 client transcript variants of AR in CRPC [38]. If variants of AR are able to maintain cellular integrity in the absence of the full-length receptor, combination therapies targeting these variants may indeed be necessary in CRPC patients.
SNX-2112 (PF-04928473) and its prodrug SNX-5422 (PF-04929113) are recently identified new-generation synthetic Hsp90 inhibitors. Both SNX compounds demonstrated superior efficacy for induction of apoptosis in LNCaP cells as compared to 17-AAG, and caused a significant delay in growth of LNCaP xenografts in mice. In addition, SNX-5422, unlike 17-AAG, prevented growth of bone metastasis [39]. This is an important characteristic of SNX-5422 that may contribute to its clinical utility.
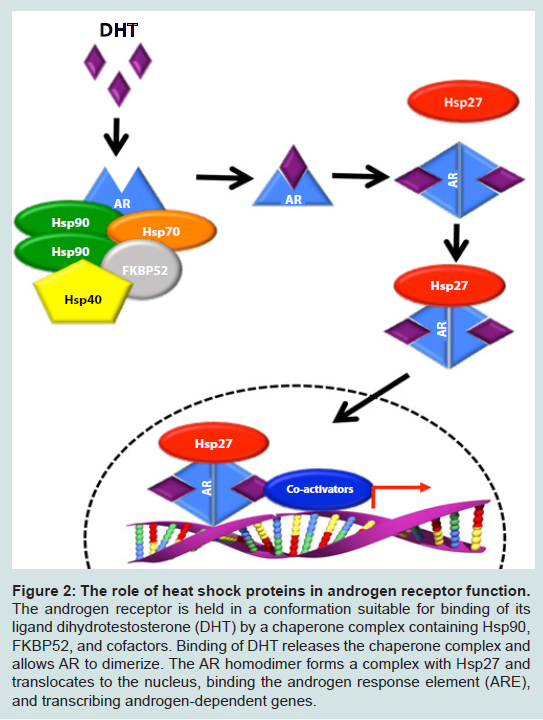
Figure 2: The role of heat shock proteins in androgen receptor function. The androgen receptor is held in a conformation suitable for binding of its ligand dihydrotestosterone (DHT) by a chaperone complex containing Hsp90, FKBP52, and cofactors. Binding of DHT releases the chaperone complex and allows AR to dimerize. The AR homodimer forms a complex with Hsp27 and translocates to the nucleus, binding the androgen response element (ARE), and transcribing androgen-dependent genes.
Novobiocin Analogs and Hsp90 C-domain Inhibitors
KU174 is a second-generation novobiocin analog. Compounds of this lineage can bind the C-terminus of Hsp90 and inhibit its activity without eliciting a heat shock response [40], which may otherwise protect cells from apoptosis. In vitro and in vivo work by Vielhauer’s and Blagg’s group showed KU174-induced Hsp90 complex degradation in PC3-MM2 and LNCaP-LN3 prostate cancer cell lines with bias toward constitutively expressed Hsp90β-containing complexes, a decrease in cell viability after KU174 treatment in PC3-MM2 cultures, and a decrease in tumor volume in rats with PC3-MM2 xenografts following KU174 treatment [41]. They further suggest that different N- or C-domain Hsp90 inhibitors are able to target distinct subsets of client proteins [41]. F-4, a second novobiocin analog, exhibited superior potency when compared to 17-AAG at equivalent concentrations. This was quantified by inhibition of proliferation and induction of apoptosis in PC3 and LNCaP cells. As with KU174, F-4 does not induce heat shock proteins [42], making these C-terminal inhibitors promising clinical candidates.Previously Evaluated Hsp90 Inhibitors in the Clinic for CRPC
Hsp90 inhibitors show promising preclinical results for CRPC treatment, but first generation inhibitors have encountered major hurdles in patient populations. Previous trials that have laid the foundation for current research are outlined in the following section (Table 1).17-AAG was used to treat fifteen prostate cancer patients with previous systemic therapy and rising PSA levels in a phase II clinical trial. Although an impressive 25% decrease in PSA levels was detected in one patient, the PSA levels in this trial were generally static and led to a halt in patient enrollment [43]. It is speculated that 17-AAG would be more effective as a drug cocktail that includes an apoptosisinducing agent to hypersensitize the cells to Hsp90 inhibition due to dose-limiting toxicity [44]. However, due to its extreme side effects, clinical trials for 17-AAG have been discontinued.
In an attempt to improve 17-AAG, the water-soluble analog 17-DMAG (alvespimycin) was developed to increase oral bioavailability. Initial phase I trials were disappointing, however a phase I study of weekly 17-DMAG in a cohort containing CRPC patients reported a complete response sustained for 124 weeks in one patient, confirmed by PSA and radiology, and progression-free survival for 59 weeks in a second patient [45]. This treated cohort also yielded one 159-week partial response in melanoma, and progression-free survival in chondrosarcoma and renal cancer for 28 and 76 weeks, respectively [45]. Despite this, 17-DMAG is not a component of any ongoing clinical trials.
IPI-504 (retaspimycin) is a hydroquinone hydrochloride salt derivative of 17-DMAG that was investigated in phase II trials for CRPC [30]. Its increased solubility (as compared to 17-AAG) was predicted to decrease adverse side effects by achieving more favorabledrug delivery.
Unfortunately there were two drug-related deaths (from ketoacidosis and hepatic failure, respectively) in the cohort containing patients with previous docetaxel treatment. However,one chemotherapy-naive patient responded positively with a 48% decrease in PSA levels [46]. This finding further encourages the idea that Hsp90 inhibition may only be a viable option for a subset of CRPC patients [47].
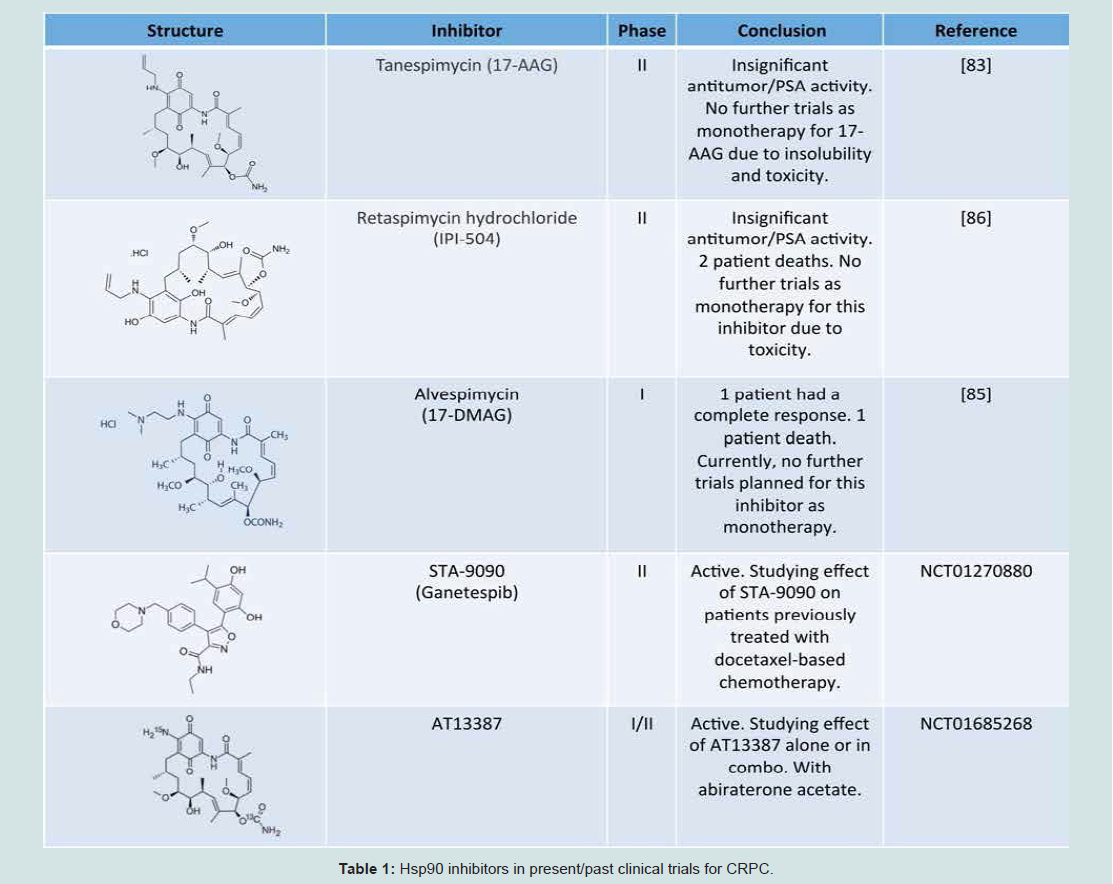
Table 1: Hsp90 inhibitors in present/past clinical trials for CRPC.
Hsp90 Inhibitors in Active Clinical Trials for CRPC
The second active trial (NCT01685268, www.clinicaltrials.gov) employs the Hsp90 inhibitor AT13387 both as a single agent and in combination with abiraterone acetate and prednisone in CRPC patients who have stopped responding to abiraterone and prednisone alone. Abiraterone decreases levels of circulating testosterone by preventing the enzymatic reactions that lead to the formation of androgens. The primary outcome measures are the safety of the dosing schedule, change in PSA and circulating tumor cell count, and change in tumor volume. Secondary outcomes are drug pharmacokinetics, PFS, and overall survival. AT13387 trials are also underway for nonsmall cell lung cancer, gastrointestinal stromal tumors, and breast cancer.
Despite the notion that Hsp90 is a viable target in prostate cancer, an important factor that bears emphasis is determining the optimum time to administer treatment. Metastatic prostate cancer frequently finds its way into the bone, a unique environment that requires consideration in this context [48]. Neckers’ group has shown that 17-AAG, though capable of effecting therapeutic response in the localized tumor, promotes the intratibial growth of PC-3M xenografts in mice. The mechanism for this aberrant growth was found to be the transient activation of the tyrosine kinase Src and Src-dependent activation of Akt [48]. Independent inhibition of osteoclast growth by three distinct inhibitors (dasatinib, bisphosphonate alendronate, and reveromycin A) abrogated this 17-AAG-induced growth [48]. Going forward, it would be sensible to monitor bone density and metastasis during therapeutic interventions in prostate cancer utilizing Hsp90 inhibitors. These data also highlight the need for a reliable clinical biomarker able to discern between localized and metastatic prostate cancer. This finding further suggests a novel combinational therapeutic avenue in the form of Hsp90 inhibition with either a Src inhibitor or specific inhibitor of osteoclast growth for the treatment of metastatic prostate cancer.
Co-inhibition of multiple targets is an emerging strategy in dealing with aggressive malignancies, with the goal of increasing efficacy by additive or synergistic effects [49,50]. It is known that Hsp90 inhibition sensitizes prostate cancer cells to chemotherapies and radiation [51,52]. Multiple instances of combination therapy in pre-clinical cell culture and animal models of prostate cancer have already been demonstrated [48]. For example, 17-AAG has been shown to synergize with inhibitors of heat shock response and proteasome inhibitors in killing PC-3 cells [53]. Further, GA and TRAIL (an activator of extrinsic apoptosis pathway) cooperatively inhibited TRAIL-resistant LNCaP cells (Table 2) [54]. These data show proof of principle for the development of combination therapies in hopes of increasing drug efficacy and avoiding the dose limiting toxicities associated with Hsp90 inhibitors in monotherapy.
Timing Hsp90 Inhibition in Prostate Cancer Patients
Combination Therapies Utilizing Hsp90 Inhibition
This strategy has been recently utilized in prostate cancer in the form of concomitant mTOR, ERK, and Hsp90 inhibition (Table 2) [55]. This co-inhibition (by rapamycin, CI-1040, and 17-AAG, respectively) causes the degradation of Slug, a transcription factor and known regulator of metastatic potential located at the confluence of these pathways. As a result, this co-inhibition caused a significant increase in apoptosis in DU145, PC3, LNCaP and 22Rv1 cell lines as well as an increase in the expression of E-cadherin, a cell-cell adhesion protein and known tumor suppressor [55].
The disruption of E-cadherin is a hallmark of early epithelial to mesenchymal transition (EMT), the transformation of cells to a metastatic state. Recent data from Isaacs’ group reports extracellular Hsp90 (eHsp90) to be a central regulator of this mechanism [56]. Expression of eHsp90 upregulates Slug which causes repression of E-cadherin transcription and an increase in matrix metalloproteinases, another marker of invasive prostate cancer [56]. Indeed, eHsp90 is more highly expressed in metastatic prostate cancer lines and eHsp90 expression is required for maintenance of metastasis, as inhibition of eHsp90 by non-permeable GA reversed EMT events in ARCaPE cells [57]. Further, Hsp90 is secreted in hypoxic exosomes by LNCaP cells, and these exosomes were shown to induce an invasive phenotype in PC3 cells [58]. It has also been shown that eHsp90 becomes hyperacetylated following histone deacetylase inhibitor treatment. This limited tumor cell invasion in T47D breast cancer cells [59], potentially making modification of eHsp90 an attractive target in mCRPC and displaying that Hsp90 is accessible via multiple modes of inhibition (Table 2).
Another co-inhibition strategy involves inhibition of Hsp90 and Hsp27. Hsp27 is a molecular chaperone upregulated during hormone therapy and via the heat shock response induced by Hsp90 inhibition. As first proposed by Neckers’ group, Hsp27 activation is secondary to Hsp90 inhibition, and as Hsp27 is involved in nuclear translocation of AR [60], this subsequent activation could effectively neutralize the action of Hsp90 inhibition (Figure 2), [61,62]. OGX-427, an antisense oligonucleotide (ASO) targeting Hsp27 is currently in Phase II clinical trials for a number of malignancies, including bladder cancer, non-small cell lung cancer, pancreatic cancer, and CRPC (clinicaltrials.gov). In combination, OGX-427 and Hsp90 inhibitors SNX-2112 (PF-04928473), SNX-5422 (PF-04929113) or 17-AAG significantly decreased tumor volume in LNCaP xenografts in mice when compared to Hsp90 inhibitors alone [63].
Gleave’s group also conducted similar experiments utilizing another ASO, OGX-011, developed to target the molecular chaperone Clusterin. OGX-011 is in Phase II/III clinical trials in combination with docetaxel and cabazitaxel for the treatment of prostate cancer and non-small cell lung cancer (clinicaltrials.gov). This chaperone is a component of the cell stress response, and was demonstratedin this study to have a role in HSF-1 regulation. Similar to Hsp27 knockdown, Clusterin silencing sensitized prostate cancer cells to Hsp90 inhibition both in vitro and in vivo (Table 2) [64]. This further confirms the model that Hsp90 inhibition-induced heat shock response is cytoprotective in tumors, and that co-inhibition may circumvent this survival mechanism.
NF-κB is a transcription factor responsible for transcribing many genes including CXCL8 and perhaps even Hsp90 [65]. Increased levels of CXCL8 (IL-8), a CXCR1/2 ligand, as well as CXCR1 and 2 are noted in prostate cancer and CXCL8 signaling is associated with progression to CRPC. Interestingly, pharmacologic inhibition of Hsp90 by GA or 17-AAG in conjunction with the CXCR2 antagonist AZ10397767 or the NF-κB inhibitor BAY11-7082 led to decreased viability of PC3 cells but not DU145 cells (Table 2). This is likely due to increased reliance of highly metastatic PC3 cells on CXCL8 signaling. As it was alluded to earlier, NF-κB inhibition causes an increase in Hsp90 mRNA, suggesting a role for NF-κB in transcriptional regulation of Hsp90 [65]. Contrasting data, as mentioned earlier, shows ganetespib to be a potent cytotoxic agent in prostate cancer cell lines regardless of AR sensitivity or metastatic potential (Table 2) [37]. This finding highlights the observed differences between first-generation and newgeneration Hsp90 inhibitors.
Furthermore, Iwai et al., and Mollapour et al., have demonstrated that inhibition of Wee1 kinase can hypersensitize PC3 cells to Hsp90 inhibitors such as 17-AAG, ganetespib, SNX-2112 and RD (Table 2) [66,67]. Wee1 is a tyrosine kinase that is an important regulator of G2/M cell cycle progression as well as an Hsp90 client [68]. Wee1 phosphorylates a conserved residue, Y24 in yeast and Y38 in human Hsp90. Interestingly, mutation of these residues to a nonphosphorylatable form (Y24F, Y38F) negatively impacts chaperoning of a subset of clients, including v-Src and HSF-1 [67].
Inhibition of Wee1 in a mouse PC3 xenograft model caused hypersensitization to Hsp90 inhibitors and activation of intrinsic apoptotic pathway [66] (Table 2). These data support the idea that combination therapy can synergize to undermine tumor growth. Mechanistically, this further suggests that in order for Hsp90 inhibition to be maximally effective, a heat shock response must be prevented.
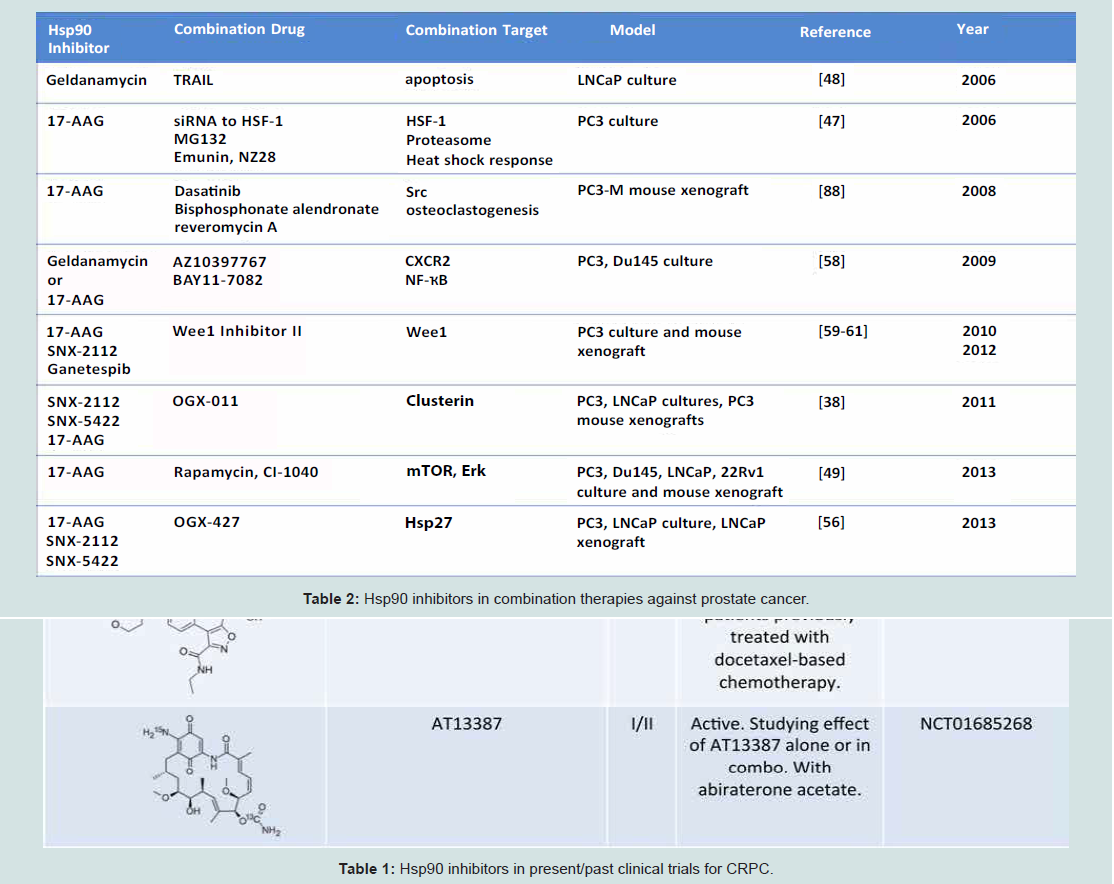
Table 2: Hsp90 inhibitors in combination therapies against prostate cancer.
Targeting the Regulators of Hsp90
Post-translational modificationsPost-translational modification of Hsp90 plays a major role in fine-tuning chaperone function [69]. Therefore targeting the enzymes that catalyze such reactions can deregulate Hsp90 function and also enhance the efficacy of Hsp90 inhibitors.
Acetylation/deacetylation is a commonly dysregulated mechanism in cancer [70]. Histone deacetylases (HDACs) are commonly overexpressed in prostate cancers, and are therapeutic targets in a number of diseases. In particular, the HDAC inhibitors Vorinostat (SAHA) and Pracinostat (SB939) are currently in multiple clinical trials against advanced solid malignancies including metastatic prostate cancer [71].
PXD101 is a potent HDAC inhibitor that has demonstrated degradation of AR and apoptosis in LNCaP, C81 and 22Rv1 cell lines and decreased tumor volume in 22Rv1 xenografts in mice [72]. HDAC inhibition, specifically that of HDAC6 has been shown to induce hyperacetylation of Hsp90, abrogate ATP-dependent chaperoning, degrade client proteins, and increase binding of Hsp90 to 17-AAG [73]. This data suggests that HDAC inhibition can prevent progression of prostate cancer to CRPC.
Romidepsin, a cyclic depsipeptide pan-HDAC inhibitor, recently entered a phase II clinical trial of prostate cancer [74]. This drug was predicted to abrogate androgen receptor function through hyperacetylation of both Hsp90 and histones. Overwhelming negative side effects including nausea, fatigue, and vomiting, led to early discontinuation of the trial for many patients. Despite this, five patients displayed regression of their PSA levels, with one patient having a cumulative decrease of 90% [74]. This further supports the idea that many current treatments are indicated only for a subset of CRPC patients and because of dose limiting toxicity, many treatments may still benefit from the synergistic effects of multiple inhibitors.
Co-Chaperones
Cdc37The co-chaperone Cdc37 is able to arrest the ATPase cycle of Hsp90 and therefore facilitate the recruitment of mainly kinase client proteins to the Hsp90 system [75,76]. Cdc37 has recently been shown to interact with Vav3, a co-activator of AR transcriptional activity [77]. This finding by Burnstein’s group demonstrates that Cdc37 potentiates Vav3 interaction with AR and increases prostate cell proliferation without affecting other characteristics of Vav3, including expression, subcellular localization, or nucleotide exchange activity [77]. It is unclear whether Cdc37 directly facilitates this process or if it occurs via interaction with Hsp90.
Workman’s group have shown recently that siRNA knockdown of Cdc37 sensitizes colon cancer cell lines to Hsp90 inhibitors, however this is due to absence of Cdc37 and not lack Cdc37-Hsp90 interaction [78]. So far, no specific inhibitors of Cdc37 have beenreported. Natural product inhibitors such as celastrol, withaferin A and the flavonoid taxifolin disrupt or are predicted to disrupt the interaction between Hsp90 and Cdc37 and show anticancer activity, [79-81] although this has not been linked directly to Cdc37 blockade, particularly in the case of celastrol, which elicits its inhibitory effect via multiple mechanisms.
p23
p23 is another Hsp90 co-chaperone implicated in the pathogenesis of prostate cancer [82]. Under normal conditions, p23 is responsible for the folding of steroid receptors in conjunction with the Hsp90 chaperone complex, but it also has Hsp90-independent functions such as prostaglandin synthesis and steroid receptor signaling [82]. Recent data revealed p23 as a bridge from AR to Hsp90 and, independently, as an activator of AR that leads to increased association of AR with androgen response element (ARE) [82]. These data conclude that co-inhibition of p23 and Hsp90 may lead to great clinical efficacy in prostate cancer.
A specific inhibitor of p23, gedunin, has already been identified and has been shown by Blagg’s group to inhibit Hsp90 chaperoning with minimal heat shock induction [27]. As well, gedunin induced apoptosis in multiple breast cancer lines and HeLa cervical cancer cells, but not non-transformed immortalized cells, showing that gedunin-mediated p23 inhibition displays specificity for malignant cells [27].
Immunophilin FKBP52
FKBP52 is an Hsp90 co-chaperone involved in stabilization of Hsp90-AR interaction (Figure 2) [83]. FKBP52 -/- MEF cells display defects in AR-dependent transcription and knockout mice present phenotypes consistent with partial androgen insensitivity [83]. Inhibition of FKBP52 by MJC13 prevented androgen-mediated dissociation of AR from the chaperone complex, and MJC13-treated LNCaP and VCaP cells displayed a significant loss of PSA expression.
This inhibition blocks nuclear translocation of AR and therefore transcription of androgen-dependent genes, evidenced by enhanced binding of AR to Hsp90 in the presence of MJC13 [84]. Interestingly, an inducible LNCaP cell line expressing V7 (an AR ligand incompetent transcript variant) demonstrates indifference to Hsp90 inhibition by GA or MJC13 [38]. Further study of this inhibitor is warranted, and perhaps in combination with Hsp90 inhibitors may provide a novel therapeutic intervention in CRPC.
SGTA
Evidence is mounting towards an important role for the recently classified co-chaperone small glutamine-rich tetratricopeptide repeat-containing protein α (SGTA) in prostate cancer [85,86].SGTA has been shown to interact with AR, and is thought to act as a tether, maintaining AR in the cytoplasm. Further, SGTA positively regulates Akt signaling, therefore aiding survival and proliferation of prostate cancer. The finding that SGTA ablation generally decreases AR mediated transcription [85] identifies this co-chaperone as a candidate for inhibition, either alone or in combination with Hsp90.
Concluding Remarks
Prostate cancer cell lines, though very powerful tools to study this disease at the molecular and cellular biology levels, are clearly incomplete models, as evidenced by the lack of translational success. One cannot be expected to accurately gauge the effect of inhibitors on a complex tumor microenvironment in a monolayer of prostate cancer cells, or be able to extrapolate the effect of inhibitors on distal metastatic sites. There appear to be many avenues to explore when it comes to choosing the optimum combination therapy, and it is likely that more than one combinational therapy will prove efficacious. These data also suggest that the recent movement toward personalized medicine will ultimately dictate therapies in prostate cancer, and the clinical answer for the vast majority of prostate cancer treatment may still lie in one of the aforementioned cooperative treatment modalities.
References
- Ferlay J, Steliarova-Foucher E, Lortet-Tieulent J, Rosso S, Coebergh JW, et al. (2013) Cancer incidence and mortality patterns in Europe: estimates for 40 countries in 2012. Eur J Cancer 49: 1374-1403.
- Siegel R, Naishadham D, Jemal A (2013) Cancer statistics, 2013. CA Cancer J Clin 63: 11-30.
- Shen MM, Abate-Shen C (2010) Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 24: 1967-2000.
- Chatterjee B (2003) The role of the androgen receptor in the development of prostatic hyperplasia and prostate cancer. Mol Cell Biochem 253: 89-101.
- Chen Y, Sawyers CL, Scher HI (2008) Targeting the androgen receptor pathway in prostate cancer. Curr Opin Pharmacol 8: 440-448.
- Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, et al. (2008) Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol 26: 242-245.
- Petrylak DP, Tangen CM, Van Veldhuizen PJ Jr, Goodwin JW, Twardowski PW, et al. (2010) Results of the Southwest Oncology Group phase II evaluation (study S0031) of ZD1839 for advanced transitional cell carcinoma of the urothelium. BJU Int 105: 317-321.
- Shirley M, McCormack PL (2014) Radium-223 Dichloride: A Review of Its Use in Patients with Castration-Resistant Prostate Cancer with Symptomatic Bone Metastases. Drugs.
- Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, et al. (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367: 1187-1197.
- de Bono JS, Kristeleit R, Tolcher A, Fong P, Pacey S, et al. (2008) Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamatehistone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res 14: 6663-6673.
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, et al. (2010) Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. NEngl J Med 363: 411-422.
- de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, et al. (2010) Prednisone plus cabazitaxel or mitoxantrone for metastatic castrationresistantprostate cancer progressing after docetaxel treatment: a randomised openlabel trial. Lancet 376: 1147-1154.
- Attard G, Cooper CS, de Bono JS (2009) Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell 16: 458-462.
- Neckers L, Workman P (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res 18: 64-76.
- Taipale M, Jarosz DF, Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11: 515-528.
- Rohl A, Rohrberg J, Buchner J (2013) The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sc 3: 253-262.
- Neckers L, Trepel JB (2013) Stressing the development of small molecules targeting Hsp90. Clin Cancer Res 20: 275-277.
- Ali MM, Roe SM, Vaughan CK, Meyer P, Panaretou B, et al. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex.Nature 440: 1013-1017.
- Shiau AK, Harris SF, Southworth DR, Agard DA (2006) Structural Analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformationalrearrangements. Cell 127: 329-340.
- Prodromou C (2012) The ‘active life’ of Hsp90 complexes. Biochim Biophys Acta 1823: 614-623.
- Li J, Soroka J, Buchner J (2012) The Hsp90 chaperone machinery: Conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta 1823: 624-635.
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, et al. (2001) The cochaperone CHIP regulates protein triage decisions mediated by heat-shockproteins. Nat Cell Biol 3: 93-96.
- Beebe K, Mollapour M, Scroggins B, Prodromou C, Xu W, et al. (2013) Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 4: 1065-1074.
- Taldone T, Sun W, Chiosis G (2009) Discovery and development of heat shock protein 90 inhibitors. Bioorg Med Chem 17: 2225-2235.
- Zhang Y, Dayalan Naidu S, Samarasinghe K, Van Hecke GC, Pheely A, et al. (2014) Sulphoxythiocarbamates modify cysteine residues in HSP90 causingdegradation of client proteins and inhibition of cancer cell proliferation. Br JCancer 110: 71-82.
- Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM (2000) The heat shock protein 90 antagonist novobiocin interacts with a previouslyunrecognized ATP-binding domain in the carboxyl terminus of the chaperone.J Biol Chem 275: 37181-37186.
- Patwardhan CA, Fauq A, Peterson LB, Miller C, Blagg BS, et al. (2013) Gedunin inactivates the co-chaperone p23 protein causing cancer cell deathby apoptosis. J Biol Chem 288: 7313-7325.
- Zhao H, Blagg BS (2013) Novobiocin analogues with second-generation noviose surrogates. Bioorg Med Chem Lett 23: 552-557.
- Travers J, Sharp S, Workman P (2012) HSP90 inhibition: two-pronged exploitation of cancer dependencies. Drug Discov Today 17: 242-252.
- Jhaveri K, Taldone T, Modi S, Chiosis G (2012) Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. BiochimBiophys Acta 1823: 742-755.
- Solit DB, Scher HI, Rosen N (2003) Hsp90 as a therapeutic target in prostate cancer. Semin Oncol 30: 709-716.
- O’Malley KJ, Langmann G, Ai J, Ramos-Garcia R, Vessella RL, et al. (2012) Hsp90 inhibitor 17-AAG inhibits progression of LuCaP35 xenograft prostate tumors to castration resistance. Prostate 72: 1117-1123.
- Saporita AJ, Ai J, Wang Z (2007) The Hsp90 inhibitor, 17-AAG, prevents the ligand independent nuclear localization of androgen receptor in refractory prostate cancer cells. Prostate 67: 509-520.
- Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, et al. (2008) NVPAUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68: 2850-2860.
- Centenera MM, Gillis JL, Hanson AR, Jindal S, Taylor RA, et al. (2012) Evidence for efficacy of new Hsp90 inhibitors revealed by ex vivo culture ofhuman prostate tumors. Clin Cancer Res 18: 3562-3570.
- Proia DA, Bates RC (2014) Ganetespib and HSP90: Translating Preclinical Hypotheses into Clinical Promise. Cancer Res 74: 1294-1300.
- He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, et al. (2013) Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. Int J Oncol 42: 35-43.
- Shafi AA, Cox MB, Weigel NL (2013) Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptoractivity and expression. Steroids 78: 548-554.
- Lamoureux F, Thomas C, Yin MJ, Kuruma H, Fazli L, et al. (2011) A novel HSP90 inhibitor delays castrate-resistant prostate cancer without alteringserum PSA levels and inhibits osteoclastogenesis. Clin Cancer Res 17: 2301-2313.
- Shelton SN, Shawgo ME, Matthews SB, Lu Y, Donnelly AC, et al. (2009) KU135, a novel novobiocin-derived C-terminal inhibitor of the 90-kDa heatshock protein, exerts potent antiproliferative effects in human leukemic cells.Mol Pharmacol 76: 1314-1322.
- Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, et al. (2011) Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 11: 468.
- Matthews SB, Vielhauer GA, Manthe CA, Chaguturu VK, Szabla K, et al. (2010) Characterization of a novel novobiocin analogue as a putative C-terminal inhibitor of heat shock protein 90 in prostate cancer cells. Prostate 70: 27-36.
- Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, et al. (2008) A phase II trial of 17- allylamino-17-demethoxygeldanamycin in patients withhormone-refractory metastatic prostate cancer. Clin Cancer Res 14: 7940-7946.
- Powers MV, Workman P (2006) Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr Relat Cancer13: S125-S135.
- Pacey S, Wilson RH, Walton M, Eatock MM, Hardcastle A, et al. (2011) A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin Cancer Res17: 1561-1570.
- Oh WK, Galsky MD, Stadler WM, Srinivas S, Chu F, et al. (2011) Multicenter phase II trial of the heat shock protein 90 inhibitor, retaspimycin hydrochloride (IPI-504), in patients with castration-resistant prostate cancer. Urology 78: 626-630.
- Xin L, Jhaveri K, Modi S (2012) HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv pharmacol 65: 471-517.
- Yano A, Tsutsumi S, Soga S, Lee MJ, Trepel J, et al. (2008) Inhibition of Hsp90 activates osteoclast c-Src signaling and promotes growth of prostatecarcinoma cells in bone. Proc Natl Acad Sci U S A 105: 15541-15546.
- Lu X, Xiao L, Wang L, Ruden DM (2012) Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitorsand other anti-cancer drugs. Biochem Pharmacol 83: 995-1004.
- Ischia J, Saad F, Gleave M (2013) The promise of heat shock protein inhibitors in the treatment of castration resistant prostate cancer. Curr opinurol 23: 194-200.
- Banerji U (2009) Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 15: 9-14.
- Gandhi N, Wild AT, Chettiar ST, Aziz K, Kato Y, et al. (2013) Novel Hsp90 inhibitor NVPAUY922 radiosensitizes prostate cancer cells. Cancer Biol Ther 14: 347-356.
- Zaarur N, Gabai VL, Porco JA, Jr., Calderwood S, Sherman MY (2006) Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res 66: 1783-1791.
- Ma Y, Lakshmikanthan V, Lewis RW, Kumar MV (2006) Sensitization of TRAIL-resistant cells by inhibition of heat shock protein 90 with low-dosegeldanamycin. Mol Cancer Ther 5: 170-178.
- Ding G, Feng C, Jiang H, Ding Q, Zhang L, et al. (2013) Combination of rapamycin, CI-1040, and 17-AAG inhibits metastatic capacity of prostate cancer via Slug inhibition. PLoS One 8: e77400.
- Hance MW, Dole K, Gopal U, Bohonowych JE, Jezierska-Drutel A, et al. (2012) Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J Biol Chem 287: 37732-37744.
- Bohonowych JE, Hance MW, Nolan KD, Defee M, Parsons C, et al. (2014) Extracellular Hsp90 mediates an NF-kappaB dependent inflammatory stromal program: Implications for the prostate tumor microenvironment. Prostate 74: 395-407.
- Ramteke A, Ting H, Agarwal C, Mateen S, Somasagara R, et al. (2013) Exosomes secreted under hypoxia enhance invasiveness and stemness ofprostate cancer cells by targeting adherens junction molecules. Molecular carcinogenesis.
- Yang Y, Rao R, Shen J, Tang Y, Fiskus W, et al. (2008) Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res 68: 4833-4842.
- Zoubeidi A, Zardan A, Beraldi E, Fazli L, Sowery R, et al. (2007) Cooperative interactions between androgen receptor (AR) and heat-shock protein 27 facilitate AR transcriptional activity. Cancer Res 67: 10455-10465.
- Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10: 537-549.
- Shiota M, Bishop JL, Nip KM, Zardan A, Takeuchi A, et al. (2013) Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 73: 3109-3119.
- Lamoureux F, Thomas C, Yin MJ, Fazli L, Zoubeidi A, et al. (2013) Suppression of Heat Shock Protein 27 Using OGX-427 Induces Endoplasmic Reticulum Stress and Potentiates Heat Shock Protein 90 Inhibitors to Delay Castrate-resistant Prostate Cancer. Eur Urol.
- Lamoureux F, Thomas C, Yin MJ, Kuruma H, Beraldi E, et al. (2011) Clusterin inhibition using OGX-011 synergistically enhances Hsp90 inhibitor activity bysuppressing the heat shock response in castrate-resistant prostate cancer.Cancer Res 71: 5838-5849.
- Seaton A, Maxwell PJ, Hill A, Gallagher R, Pettigrew J, et al. (2009) Inhibition of constitutive and cxc-chemokine-induced NF-kappaB activity potentiatesansamycin-based HSP90-inhibitor cytotoxicity in castrate-resistant prostate cancer cells. Br J Cancer 101: 1620-1629.
- Iwai A, Bourboulia D, Mollapour M, Jensen-Taubman S, Lee S, et al. (2012) Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 11: 3649-3655.
- Mollapour M, Tsutsumi S, Donnelly AC, Beebe K, Tokita MJ, et al. (2010) Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinctfacets of chaperone function. Mol Cell 37: 333-343.
- Mollapour M, Tsutsumi S, Neckers L (2010) Hsp90 phosphorylation, Wee1 and the cell cycle. Cell Cycle 9: 2310-2316.
- Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, et al. (2013) Contributions of cochaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Med Chem 5:1059-1071.
- Zismanov V, Drucker L, Gottfried M (2013) ER homeostasis and motility of NSCLC cell lines can be therapeutically targeted with combined Hsp90 andHDAC inhibitors. Pulm Pharmcol & Ther 26: 388-394.
- Ramaswamy B, Fiskus W, Cohen B, Pellegrino C, Hershman DL, et al. (2012)Phase I-II study of vorinostat plus paclitaxel and bevacizumab in metastatic breast cancer: evidence for vorinostat-induced tubulin acetylation and Hsp90inhibition in vivo. Breast Cancer Res Treat. 132:1063-1072.
- Gravina GL, Marampon F, Muzi P, Mancini A, Piccolella M, et al. (2013) PXD101 potentiates hormonal therapy and prevents the onset of castrationresistant phenotype modulating androgen receptor, HSP90, and CRM1 in preclinical models of prostate cancer. Endocr Relat Cancer 20: 321-337.
- Rao R, Fiskus W, Yang Y, Lee P, Joshi R, et al. (2008) HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 112: 1886-1893.
- Molife LR, Attard G, Fong PC, Karavasilis V, Reid AH, et al. (2010) Phase II, two-stage, single arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration resistant prostate cancer (CRPC). Ann Oncol 21:109-113.
- Siligardi G, Panaretou B, Meyer P, Singh S, Woolfson DN, et al. (2002) Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37.J Biol Chem 277: 20151-20159.
- Abbas-Terki T, Briand PA, Donze O, Picard D (2002) The Hsp90 cochaperones Cdc37 and Sti1 interact physically and genetically. Biol Chem383: 1335-1342.
- Wu F, Peacock SO, Rao S, Lemmon SK, Burnstein KL (2013) Novel interaction between the co-chaperone Cdc37 and Rho GTPase exchange factor Vav3 promotes androgen receptor activity and prostate cancer growth.J Biol Chem 288: 5463-5474.
- Smith JR, de Billy E, Hobbs S, Powers M, Prodromou C, et al. (2013) Restricting direct interaction of CDC37 with HSP90 does not compromise chaperoning of client proteins. Oncogene.
- Zhang T, Li Y, Yu Y, Zou P, Jiang Y, et al. (2009) Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 284: 35381-35389.
- Yu Y, Hamza A, Zhang T, Gu M, Zou P, et al. (2010) Withaferin A targets heat shock protein 90 in pancreatic cancer cells. Biochem Pharmacol 79: 542-551.
- Verma S, Singh A, Mishra A (2012) Dual inhibition of chaperoning process by taxifolin: molecular dynamics simulation study. J Mol Graph Model 37: 27-38.
- Reebye V, Querol Cano L, Lavery DN, Brooke GN, et al. (2012) Role of the HSP90-associated cochaperone p23 in enhancing activity of the androgen receptor and significance for prostate cancer. Mol Endocrinol 26: 1694-1706.
- Cano LQ, Lavery DN, Bevan CL (2013) Mini-review: Foldosome regulation of androgen receptor action in prostate cancer. Mol Cell Endocrinol 369: 52-62.
- De Leon JT, Iwai A, Feau C, Garcia Y, Balsiger HA, et al. (2011) Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc Natl Acad Sci U S A 108: 11878-11883.
- Trotta AP, Need EF, Selth LA, Chopra S, Pinnock CB, et al. (2013) Knockdown of the cochaperone SGTA results in the suppression of androgen and PI3K/Akt signaling and inhibition of prostate cancer cell proliferation. Int J Cancer133: 2812-2823.
- Philp LK, Butler MS, Hickey TE, Butler LM, Tilley WD, et al. (2013) SGTA: a new player in the molecular co-chaperone game. Hormon Cancer 46: 343-357.