
Journal of Vaccine & Immunotechnology
Download PDF
Research Article
*Address for Correspondence: Yanjin Zhang, Molecular Virology Laboratory, VA-MD Regional College of Veterinary Medicine, University of Maryland, 8075 Greenmead Drive, College Park, MD 20742,USA, Tel: +1 301 314-6596; Fax: +1 301 314-6855; E-mail: zhangyj@umd.edu
Citation: Luo A, Yang Y, Yu Y, Xiao Y, Zhang Y. Enhancing Solubilization of Hydrophobic ORF3 Product of Hepatitis E Virus in Bacterial Expression System. J Vaccine Immunotechnology. 2014;1(1): 5.
Copyright © 2013 Zhang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 10 April 2014 | Accepted: 17 June 2014 | Published: 20 June 2014
Reviewed & Approved by: Dr. Guey Chuen Perng, Department of Microbiology and Immunology, College of Medicine, National Cheng Kung University, Taiwan
Enhancing Solubilization of Hydrophobic ORF3 Product of Hepatitis E Virus in Bacterial Expression System
Allyson Luo1‡, Yonglin Yang1,2‡, Ying Yu1, Yueqiang Xiao1 and Yanjin Zhang1*
- 1Molecular Virology Laboratory, VA-MD Regional College of Veterinary Medicine and Maryland Pathogen Research Institute, University of Maryland, College Park, MD 20742
- 2Nanjing Red Cross Blood Center, Najing, Jiangsu, China
- ‡Co-first authors
*Address for Correspondence: Yanjin Zhang, Molecular Virology Laboratory, VA-MD Regional College of Veterinary Medicine, University of Maryland, 8075 Greenmead Drive, College Park, MD 20742,USA, Tel: +1 301 314-6596; Fax: +1 301 314-6855; E-mail: zhangyj@umd.edu
Citation: Luo A, Yang Y, Yu Y, Xiao Y, Zhang Y. Enhancing Solubilization of Hydrophobic ORF3 Product of Hepatitis E Virus in Bacterial Expression System. J Vaccine Immunotechnology. 2014;1(1): 5.
Copyright © 2013 Zhang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Vaccine & Immunotechnology | Volume: 1, Issue: 1
Submission: 10 April 2014 | Accepted: 17 June 2014 | Published: 20 June 2014
Reviewed & Approved by: Dr. Guey Chuen Perng, Department of Microbiology and Immunology, College of Medicine, National Cheng Kung University, Taiwan
Abstract
Hepatitis E Virus (HEV) is a major cause of acute clinical hepatitis in developing countries. HEV genome has three open reading frames: ORF1, ORF2 and ORF3. ORF3 encodes a small 13 kDa protein (hereinafter called vp13). This protein has been suggested to have multiple functions, including interference with cell signaling pathways, viral particle release from infected cells, and viral pathogenesis. However, the structural basis and mechanism by which the vp13 exerts these multiple biological functions is unclear. Characterization of vp13 has been hindered by the difficulty in efficiently producing enough hand pure vp13 proteins. This may attribute to vp13’s hydrophobic and insoluble nature. The objective of this study was to express vp13 in prokaryotic expression system and optimize the conditions to increase the soluble portion for vp13 purification. The ORF3 sequence was cloned into a bacterial expression plasmid with maltose binding protein (MBP) as a tag. The recombinant plasmid was transformed into E. coli BL-21 strain for protein expression. Most MBP-vp13 protein was found in insoluble portion, making protein purification a challenge. To increase the solubility of this recombinant protein, we optimized the induction and processing conditions. Induction at 18°C and addition of 1% glucose to the bacterium culture successfully led to efficient expression of soluble recombinant MBP-vp13 fusion protein. Further, the soluble fusion protein was easy to be purified. This enhanced solubilization and purification of vp13 protein should facilitate further study of vp13’s structure and function. This study provides an example for expression and purification of hydrophobic proteins for vaccine or immunological reagent production.Introduction
Hepatitis E virus; HEV; ORF3; vp13; Purification; Solubilization; Hydrophobic proteinIntroduction
Hepatitis E Virus (HEV) is a major cause of acute hepatitis, prevalent in developing countries [1]. Currently, one third of the world’s population is at risk of infection of the Hepatitis E Virus. An estimated fourteen million symptomatic cases of HEV infection with three hundred thousand deaths are reported annually throughout the world, primarily in developing countries including the Indian subcontinents, Southeast and Central Asia, the Middle East, and Northern/Western parts of Africa [2]. Although HEV infection has emerged as a global health threat, its relative absence in the Western world contributes to its lack of recognition. Clinical manifestation of HEV infection can range from asymptomatic to fulminant hepatitis and death. Approximately 20% of infections are symptomatic in high disease endemic areas. The death rate among pregnant women who express symptoms can reach up to 25% and it usually occurs in their third trimester [1]. HEV infection is also problematic for immunecompromised individuals and organ transplant patients [3].HEV is a small, non-enveloped, single-stranded positivesense RNA virus. The genome is approximately 7.2 kb, and has a 5’ cap and a 3’-poly A tail [4]. The virus classified as a Hepevirus in the family Hepeviridae has four genotypes yet only one serotype. Genotypes 1 and 2 infect solely humans, while genotypes 3 and 4 also infect pigs and other mammalian species [5]. The RNA genome of HEV is organized into three open reading frames (ORFs), with the nonstructural proteins encoded at the 5’ end of the genome and the structural proteins at the 3’ end. ORF1 encodes the nonstructural polyprotein; ORF2 encodes the major capsid protein; ORF3 encodes a small protein, which is 114 amino acids long [6,7]. Exact function of vp13 encoded by ORF3 is not clear, although it has been suggested that vp13 has multiple functions including interference of cell signaling pathways, viral particle release from infected cells and viral pathogenesis [5]. Some studies suggested that the expression of the intact ORF3 protein is required for the infection in vivo [6,8]. However, other studies showed that the protein is not required for infection and virion morphogenesis in vitro [9]. HEV vp13 protein has been suggested to interact with more than 30 human cellular proteins [10,11]. How such a small viral protein plays so many roles in the virus life cycle and interacts with an abundance of cellular proteins is not well understood. The structure of vp13 protein is not known. It is important to delineate the structural and functional relationship of vp3 protein. However, characterization of vp13 has been hindered by the difficulty in efficiently generating a recombinant vp13 due to the protein’s hydrophobic nature and insolubility.
The 114 amino acids of vp13 encoded by ORF3 are highly hydrophobic with 41 of the 114 amino acids are hydrophobic amino acids. The objective of this study was to enhance the expression and solubility of vp13. The hypothesis was that a recombinant fusion protein between the ORF3 protein and a maltose binding protein (MBP) would enhance the solubility of vp13 and that the induction conditions could be optimized to enhance the solubility of the recombinant protein. The induction conditions to be adjusted included relevant conditions such as IPTG concentration, induction time, and addition of glucose. The results showed that the recombinant MBP-vp13 fusion protein was successfully expressed as a soluble form that was detected by a rabbit antibody specifically against vp13. Furthermore, the recombinant MBP-vp13 fusion protein was purified by a kit specifically designed for MBP fusion protein purification. The results of this study can be extrapolated to other studies on vaccine or immunological reagent production using hydrophobic proteins.
Materials and Methods
Plasmid constructionHEV ORF3 was amplified from a pSK-E2 plasmid that contains the full-length cDNA of the HEV genome (GenBank accession no. AF444002) [12]. PCR was conducted using the primers H3F14 (5’-GGAATTCCATATGGGTTCGCGACCATGCG-3’) and H3R16 (5’-CGGCTCGAGTTAGCGGCGCGGCCCCAGCTGTG-3’), which contain restriction sites for NdeI and XhoI to facilitate directional cloning. The PCR amplified ORF3 fragment was digested with NdeI and XhoI and cloned into the expression plasmid pVL-847 [13], which is a prokaryotic expression plasmid containing MBP coding sequence. The resulting plasmid was transformed into E. coli DH5α competent cells. Miniprep DNA was prepared from the E. coli clones; positive clones were identified with digestion of the DNA with NdeI and XhoI. The integrity of positive clones was further confirmed by DNA sequencing. The glycerol stock of the positive E. coli clones was stored in a -80°C freezer until use.
Recombinant MBP-vp13 fusion protein expression
The recombinant plasmid DNA was transformed into BL21 competent cells for protein expression. The transformed BL21 cells were cultured at a 37°C shaker for overnight at 220 rpm. The next day, a fresh 2 ml LB broth was inoculated with the overnight culture at 1:500 dilution and cultured for 2-3 hours at 250 rpm and 37°C. To induce fusion protein expression, IPTG at indicated concentrations (from 0.2 to 1.0 mM) was added to the LB broth when the OD600 of the bacteria culture reaches 0.8 and incubated further in the shaker for 3 hours. The bacteria culture was spun down for 5 minutes at 10,000 xg to pellet the E. coli. The pellet was resuspended in 100 μL of PBS pH7.2 with 5 mM of EDTA in 1.5 ml microfuge tubes and sonicated for indicated time in cycles of 10 second sonication (output voltage 1000 V rms and frequency 20 KHz) and 10 second pause in a 3” diameter cup horn (Model Q500, Qsonica Sonicator Ultrasonic Processor, ewtown, CT). The sonicated samples were spun down for 10 minutes at 4°C and 14,000 xg. Both the supernatant and pellet from the sonicated samples were run on SDS-PAGE (Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis), followed by Western Blot analysis.
To detect vp13 expression, the Western Blot membrane was probed with rabbit antibody specifically against vp13 as described previously [11] or MBP monoclonal antibody (Rockland Immunochemicals, Inc., Gilbertsville, PA). The signal was detected by goat anti-rabbit or anti-mouse IgGsecondary antibody conjugated with horseradish peroxidase (Sigma-Aldrich, St. Louis, MO) and revealed using a chemiluminescence substrate. The chemiluminescence signal was recorded digitally using a ChemiDoc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Biotechnology, Inc., Dallas, TX) was also used to probe some blotting membranes for loading control.
Purification of MBP-vp13 fusion protein
To purify MBP-vp13 fusion protein, syringe- operated MBPTrap HP columns were used (GE Healthcare Life Sciences, Pittsburgh, PA) by following the manufacturer’s instructions. The syringe was filled with binding buffer (20 mMTris-HCl, 200 mMNaCl, 1 mM EDTA, pH7.4). The ethanol in the column was washed out with 5 column volumes (CV) of the binding buffer. Then, the column was equilibrated with 5 CV of binding buffer at a speed of 1 ml/min. The sample containing the fusion protein was loaded to the column by a syringe. The column was washed with 5 CV of the binding buffer to wash out unbound non-specific proteins. Finally, the protein bound to the column was eluted with 5 CV of elution buffer (binding buffer plus 10 mM maltose). The eluted fractions were combined and concentrated up to 20-fold using Amicon Ultra-4 Centrifugal Filter Unit (Millipore, Billerica, MA). Examination for the presence of MBP-vp13 was done by SDS-PAGE and Coomassie blue (Bio-Rad) staining analysis, and Western blotting with antibodies against MBP and vp13.
Results
Construction of prokaryotic expression plasmid encoding recombinant MBP-vp13 fusion proteinAmong 114 vp13 amino acids, 41 (36%) are hydrophobic.HEV vp13 was analyzed by a hydrophobicity prediction program available in the public domain (http://web.expasy.org/protparam/). The hydrophobicity index for vp13 is +0.378, suggesting the high hydrophobic nature of vp13. The hydrophobicity index is 0.972 for the first half of the protein and -0.216 for the second half, indicating most hydrophobicity is due to the N-terminal half. This may explain the difficulty in efficiently expressing a soluble full length vp13. MBP has been successfully used as a fusion tag to express hydrophobic and insoluble proteins [13]. In this study, vp13 was expressed as a fusion protein with MBP by ligating the cDNA encoding vp13 that was PCR amplified from pSK-E2 and cloned into pVL-847, which already contained the MBP coding sequence. The integrity of the expression construct was confirmed by restriction enzyme digest and direct DNA sequencing.
Majority of recombinant MBP-vp13 fusion protein in E. coli are insoluble
The fusion protein expression plasmid was transformed into BL21 competent E. coli. IPTG 1 mM was used to induce the protein expression. Whole protein from the bacterial cells was subjected to SDS-PAGE and Western blotting with vp13 antibody to check the protein expression. Among seven colonies selected, two had high level expression of MBP-vp13 protein (Figure 1).
 Figure 1: Induction of MBP-vp13 expression in E. coli at 37°C with IPTG 1 mM for 3 hours. Seven colonies were selected to inoculate LB at 2 ml each and cultured for IPTG induction. Colony 1 without induction was included as a control. The bacteria were collected and pellets were dissolved in Laemmli sample buffer. The total proteins were separated in SDS-PAGE and blotted with rabbit anti-vp13 antibody.
Figure 1: Induction of MBP-vp13 expression in E. coli at 37°C with IPTG 1 mM for 3 hours. Seven colonies were selected to inoculate LB at 2 ml each and cultured for IPTG induction. Colony 1 without induction was included as a control. The bacteria were collected and pellets were dissolved in Laemmli sample buffer. The total proteins were separated in SDS-PAGE and blotted with rabbit anti-vp13 antibody.
Knowing the fusion protein was expressed in E. coli, we checked for its solubility. After IPTG induction, the E. coli pellet was sonicated to release the protein. After sonication, the sample was separated by centrifugation into soluble fraction (supernatant) and insoluble fraction (pellet). Both fractions were run on SDS-PAGE followed by Western Blot analysis with anti-vp13 specific antibody. Surprisingly, the majority of the fusion protein, which reacted with the anti-vp13 specific antibody, was in the insoluble fraction, only a minor fraction of the target protein was detected in the soluble fraction (Figure 2A).
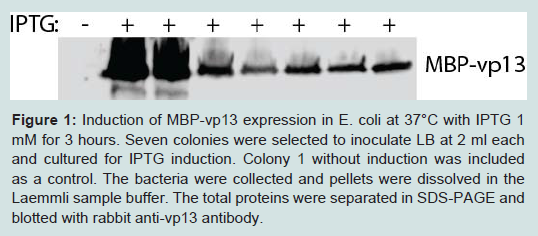 Figure 1: Induction of MBP-vp13 expression in E. coli at 37°C with IPTG 1 mM for 3 hours. Seven colonies were selected to inoculate LB at 2 ml each and cultured for IPTG induction. Colony 1 without induction was included as a control. The bacteria were collected and pellets were dissolved in Laemmli sample buffer. The total proteins were separated in SDS-PAGE and blotted with rabbit anti-vp13 antibody.
Figure 1: Induction of MBP-vp13 expression in E. coli at 37°C with IPTG 1 mM for 3 hours. Seven colonies were selected to inoculate LB at 2 ml each and cultured for IPTG induction. Colony 1 without induction was included as a control. The bacteria were collected and pellets were dissolved in Laemmli sample buffer. The total proteins were separated in SDS-PAGE and blotted with rabbit anti-vp13 antibody.
 Figure 2: Most of MBP-vp13 protein expressed is in insoluble portion: A. Detection of MBP-vp13 in supernatant and pellet after sonication and centrifugation. Four colonies were selected to inoculate LB culture, cultured at 37°C, and induced with 1 mM IPTG for 3 h. Pellets were resuspended in PBS and sonicated. The samples were centrifuged to separate supernatant and pellet. Western blotting with rabbit anti-vp13 antibody was conducted.B. Different IPTG concentrations in induction tested. Supernatant samples were used for the Western blotting with antibodies against vp13 and GAPDH. IPTG concentrations at 0 (control lane), 0.2, 0.4, 0.6, 0.8, and 1.0 mM were tested. The bacteria samples at equal volumes from different concentrations of IPTG were sonicated and analyzed by SDS-PAGE and Western blotting. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 after normalization with GAPDH are shown below the images.
Figure 2: Most of MBP-vp13 protein expressed is in insoluble portion: A. Detection of MBP-vp13 in supernatant and pellet after sonication and centrifugation. Four colonies were selected to inoculate LB culture, cultured at 37°C, and induced with 1 mM IPTG for 3 h. Pellets were resuspended in PBS and sonicated. The samples were centrifuged to separate supernatant and pellet. Western blotting with rabbit anti-vp13 antibody was conducted.B. Different IPTG concentrations in induction tested. Supernatant samples were used for the Western blotting with antibodies against vp13 and GAPDH. IPTG concentrations at 0 (control lane), 0.2, 0.4, 0.6, 0.8, and 1.0 mM were tested. The bacteria samples at equal volumes from different concentrations of IPTG were sonicated and analyzed by SDS-PAGE and Western blotting. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 after normalization with GAPDH are shown below the images.
Optimization of recombinant MBP-vp13 fusion protein expression
 Figure 4: Optimization of sonication time for vp13 release. Western blotting was done with antibodies against vp13 (A) and MBP (B). The bacteria were cultured at 18ᵒC and and 180 rpm, and induced with IPTG 0.2 mM. Total time in minutes of sonication is listed above the image. Sonication was done as cycles of 10 seconds pulse and 10 seconds pause. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Figure 4: Optimization of sonication time for vp13 release. Western blotting was done with antibodies against vp13 (A) and MBP (B). The bacteria were cultured at 18ᵒC and and 180 rpm, and induced with IPTG 0.2 mM. Total time in minutes of sonication is listed above the image. Sonication was done as cycles of 10 seconds pulse and 10 seconds pause. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Enhancing solubility of MBP-vp13 fusion protein
 Figure 5: Effect of glucose and ethanol in induction of MBP-vp13 expression as soluble protein. The bacteria were cultured at 18°C and 180 rpm, and induced with IPTG 0.2 mM for 3 h. Western blotting was done with antibodies against vp13.Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Figure 5: Effect of glucose and ethanol in induction of MBP-vp13 expression as soluble protein. The bacteria were cultured at 18°C and 180 rpm, and induced with IPTG 0.2 mM for 3 h. Western blotting was done with antibodies against vp13.Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Purification of recombinant MBP-vp13 fusion protein

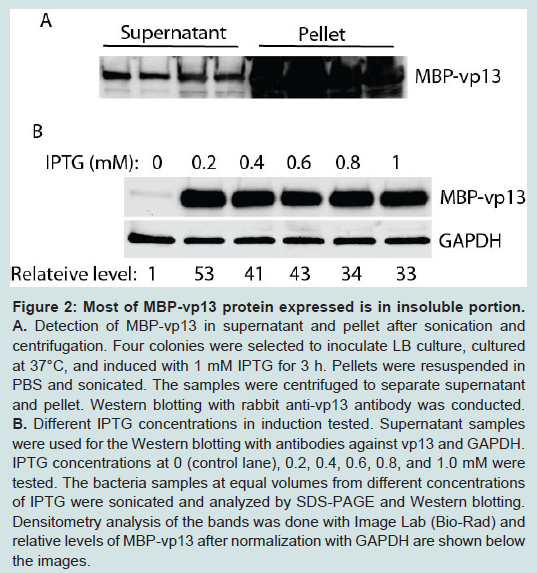 Figure 2: Most of MBP-vp13 protein expressed is in insoluble portion: A. Detection of MBP-vp13 in supernatant and pellet after sonication and centrifugation. Four colonies were selected to inoculate LB culture, cultured at 37°C, and induced with 1 mM IPTG for 3 h. Pellets were resuspended in PBS and sonicated. The samples were centrifuged to separate supernatant and pellet. Western blotting with rabbit anti-vp13 antibody was conducted.B. Different IPTG concentrations in induction tested. Supernatant samples were used for the Western blotting with antibodies against vp13 and GAPDH. IPTG concentrations at 0 (control lane), 0.2, 0.4, 0.6, 0.8, and 1.0 mM were tested. The bacteria samples at equal volumes from different concentrations of IPTG were sonicated and analyzed by SDS-PAGE and Western blotting. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 after normalization with GAPDH are shown below the images.
Figure 2: Most of MBP-vp13 protein expressed is in insoluble portion: A. Detection of MBP-vp13 in supernatant and pellet after sonication and centrifugation. Four colonies were selected to inoculate LB culture, cultured at 37°C, and induced with 1 mM IPTG for 3 h. Pellets were resuspended in PBS and sonicated. The samples were centrifuged to separate supernatant and pellet. Western blotting with rabbit anti-vp13 antibody was conducted.B. Different IPTG concentrations in induction tested. Supernatant samples were used for the Western blotting with antibodies against vp13 and GAPDH. IPTG concentrations at 0 (control lane), 0.2, 0.4, 0.6, 0.8, and 1.0 mM were tested. The bacteria samples at equal volumes from different concentrations of IPTG were sonicated and analyzed by SDS-PAGE and Western blotting. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 after normalization with GAPDH are shown below the images.To enhance the protein expression level and to increase its solubility, several conditions were tested, including concentration of IPTG, induction time and temperature. IPTG concentration from 0.2 to 1.0 mM was first tested (Figure 2B). It was found that IPTG at 0.2 mM induced 53-fold increase in MBP-vp13 fusion protein expression when compared to mock-induction control, after normalization with GAPDH. Therefore, 0.2 mM IPTG was chosen for further studies.
Next, different induction temperatures were tested. It was found that the protein can be expressed in culture at 18°C and there was minimum difference in solubility of the protein, ranging in relative levels of vp13 from 1 to 1.4-fold (Figure 3A). Majority of the vp13 fusion protein was still in pellet. So 18°C was selected for further studies to enhance the protein solubility. Induction time was then tested to find the optimal time of induction for the protein expression (Figure 3B). The protein level was similar from 1.5 h to 5.5 h at 18°C with 0.2 mM IPTG induction. Thus, three hour induction was selected for further studies of this protein.
 Figure 3: Optimization of induction temperature and time for MBP-vp13 expression: A. Effect of temperature on vp13 solubility. The bacterium culture was induced with 0.2 mM IPTG for 3 h. Western blotting with rabbit anti-vp13 antibody was conducted. Densitometry analysis of the bands was done and relative levels of MBP-vp13 are shown below the image. B. Effect of induction time of MBP-vp13 solubility in BL21 cells at IPTG 0.2 mM, 180 rpm and 18°C. Western blotting with antibody against vp13 was conducted.
Figure 3: Optimization of induction temperature and time for MBP-vp13 expression: A. Effect of temperature on vp13 solubility. The bacterium culture was induced with 0.2 mM IPTG for 3 h. Western blotting with rabbit anti-vp13 antibody was conducted. Densitometry analysis of the bands was done and relative levels of MBP-vp13 are shown below the image. B. Effect of induction time of MBP-vp13 solubility in BL21 cells at IPTG 0.2 mM, 180 rpm and 18°C. Western blotting with antibody against vp13 was conducted.
Optimization of MBP-vp13 fusion protein extraction from E. coli
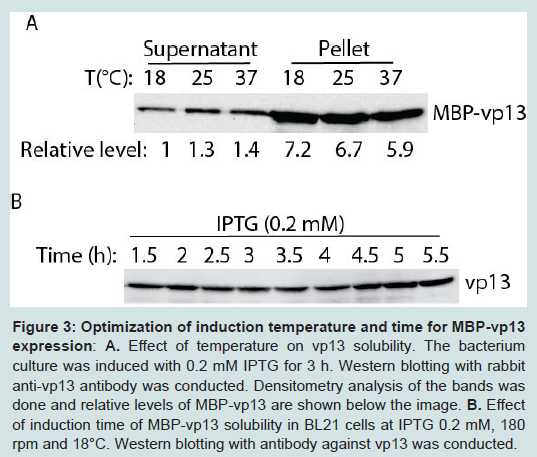 Figure 3: Optimization of induction temperature and time for MBP-vp13 expression: A. Effect of temperature on vp13 solubility. The bacterium culture was induced with 0.2 mM IPTG for 3 h. Western blotting with rabbit anti-vp13 antibody was conducted. Densitometry analysis of the bands was done and relative levels of MBP-vp13 are shown below the image. B. Effect of induction time of MBP-vp13 solubility in BL21 cells at IPTG 0.2 mM, 180 rpm and 18°C. Western blotting with antibody against vp13 was conducted.
Figure 3: Optimization of induction temperature and time for MBP-vp13 expression: A. Effect of temperature on vp13 solubility. The bacterium culture was induced with 0.2 mM IPTG for 3 h. Western blotting with rabbit anti-vp13 antibody was conducted. Densitometry analysis of the bands was done and relative levels of MBP-vp13 are shown below the image. B. Effect of induction time of MBP-vp13 solubility in BL21 cells at IPTG 0.2 mM, 180 rpm and 18°C. Western blotting with antibody against vp13 was conducted.To increase the level of MBP-vp13 in the soluble fraction, time duration of sonication was tested. At a constant power, duration of sonication ranged from 1 to 8 minutes in cycles of 10 second sonication and 10 second pause. Along with the increase of sonication time, MBP-vp13 level in the soluble fraction significantly increased while the protein level in the insoluble fraction decreased (Figure 4A, 4B). In vp13 blotting, the relative levels of MBP-vp13 in supernatant were 1 to 2.5 for the duration of sonication from 1 to 8 minutes (Figure 4A). In MBP blotting, the relative levels of MBPvp13 in supernatant were 1 to 2.3 for the duration of sonication from 1 to 8 minutes (Figure 4B). It appeared that 3-minute sonication was enough to cause release of most soluble MBP-vp13, especially shown in blotting with MBP antibody (Figure 4B). The result suggests that the duration of sonication time needs to be optimized to increase the release of the protein into the soluble fraction.
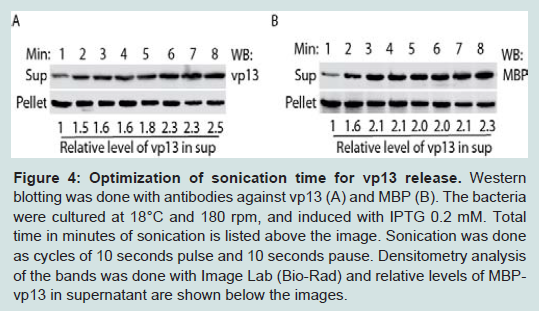 Figure 4: Optimization of sonication time for vp13 release. Western blotting was done with antibodies against vp13 (A) and MBP (B). The bacteria were cultured at 18ᵒC and and 180 rpm, and induced with IPTG 0.2 mM. Total time in minutes of sonication is listed above the image. Sonication was done as cycles of 10 seconds pulse and 10 seconds pause. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Figure 4: Optimization of sonication time for vp13 release. Western blotting was done with antibodies against vp13 (A) and MBP (B). The bacteria were cultured at 18ᵒC and and 180 rpm, and induced with IPTG 0.2 mM. Total time in minutes of sonication is listed above the image. Sonication was done as cycles of 10 seconds pulse and 10 seconds pause. Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.Glucose and ethanol were tested for their effect on overall protein expression with particular focus on the level of protein in the soluble fraction. Glucose represses induction of the lac promoter by lactose, which is present in most rich media including LB [14]. Addition of glucose at 1% and ethanol at 3% significantly increased the portion of MBP-vp13 in the soluble fraction to 2 and 1.5-fold, respectively (Figure 5). Addition of ethanol at 5% caused reduction of the fusion protein level, indicating a delicate concentration range is hard to optimize and its use should be avoided in this case. So 1% glucose was selected for further work on MBP-vp13 expression and purification.
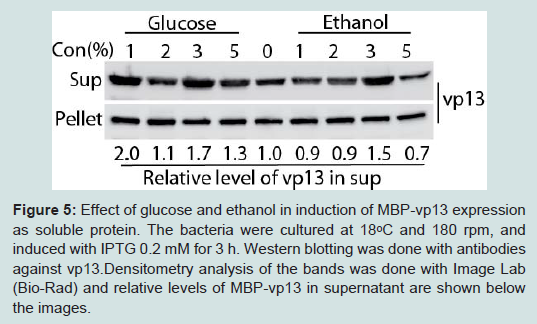 Figure 5: Effect of glucose and ethanol in induction of MBP-vp13 expression as soluble protein. The bacteria were cultured at 18°C and 180 rpm, and induced with IPTG 0.2 mM for 3 h. Western blotting was done with antibodies against vp13.Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.
Figure 5: Effect of glucose and ethanol in induction of MBP-vp13 expression as soluble protein. The bacteria were cultured at 18°C and 180 rpm, and induced with IPTG 0.2 mM for 3 h. Western blotting was done with antibodies against vp13.Densitometry analysis of the bands was done with Image Lab (Bio-Rad) and relative levels of MBP-vp13 in supernatant are shown below the images.To purify the fusion protein, a GE MBPTrap HP column was used by following the manufacturer’s instruction. The soluble fraction after sonication of the bacteria containing the fusion protein was applied to the column. After washing away the unbound non-specific proteins, the fusion protein was eluted. The eluted proteins were examined by SDS-PAGE and Coomassie blue staining. A band at ~55 kDa and some minor bands below it appeared in the gel (Figure 6A). The size of the band matches the expected size of recombinant MBP-vp13 fusion protein, indicating that the elusions contains the soluble MBP-vp13 and that purification by this simple one-step method appeared to be pure as only single major band was observed. To confirm the specificity of the band, we ran Western blot analysis with antibodies against both vp13 and MBP, showing a single major band (Figure 6B). The data suggested that MBP-vp13 fusion protein can be readily purified by the column and that the process from expression to extraction yields intact MBP-vp13 fusion protein. Densitometry analysis of the images was done and approximate calculations were conducted taking into consideration of loading amount and total volume of each sample in the purification process. It appeared that the amount of MBP-vp13 in preloading soluble portion is 27% of total MBP-vp13 in the whole culture harvested (Figure 6c), which indicates that at least more than a quarter of the protein was release into soluble portion in the initial process. The soluble vp13 portion was significantly higher than the approximate 7% in the initial optimization (Figure 3A). The total amount of eluted MBP-vp13 is 44% of the total soluble vp13 in the preloading sample. The results suggest that the purification process is efficient, yet further optimization is needed to improve recovery by avoiding overloading the MBPTrap HP column.
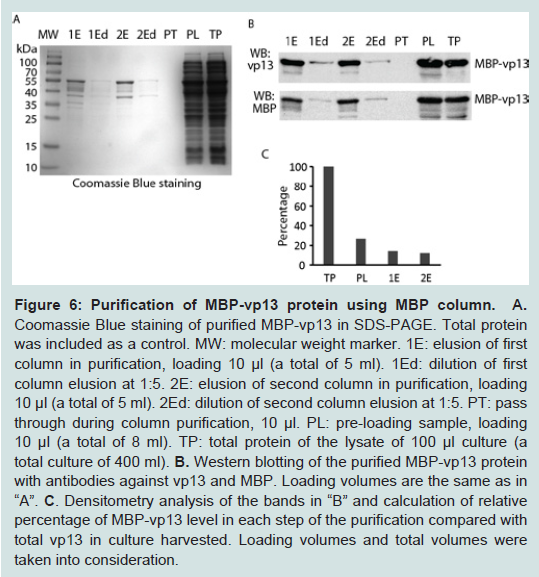
Figure 6: Purification of MBP-vp13 protein using MBP column A. Coomassie Blue staining of purified MBP-vp13 in SDS-PAGE. Total protein was included as a control. MW: molecular weight marker. 1E: elusion of first column in purification, loading 10 µl (a total of 5 ml). 1Ed: dilution of first column elusion at 1:5. 2E: elusion of second column in purification, loading 10 µl (a total of 5 ml). 1Ed: dilution of second column elusion at 1:5. PT: pass through during column purification, 10 µl. PL: pre-loading sample, loading 10 µl (a total of 8 ml). TP: total protein of the lysate of 100 µl culture (a total culture of 400 ml). B. Western blotting of the purified MBP-vp13 protein with antibodies against vp13 and MBP. Loading volumes are the same as in “A”. C. Densitometry analysis of the bands in “B” and calculation of relative percentage of MBP-vp13 level in each step of the purification compared with total vp13 in culture harvested. Loading volumes and total volumes were taken into consideration.
Discussion
Characterization and understanding of vp13 have been hindered by the difficulty in efficiently generating a soluble recombinant vp13. This could be due to the fact that vp13 is a highly hydrophobic and insoluble protein. In this study, the expression of recombinant MBP-vp13 fusion protein was optimized, including IPTG level and glucose addition. The fusion protein was efficiently extracted to increase soluble portion. A significant portion of the fusion protein was present in the soluble rather than the insoluble fraction after the optimizations. The intact fusion protein was successfully purified by a simple one-step affinity purification process.In order to study biological function of vp13 as well as its structure and function relationship, a cDNA encoding a fusion protein between maltose binding protein (MBP) and vp13 was engineered. The reason MBP was chosen as a fusion partner for vp13 was due to MBP’s hydrophilic nature. Furthermore, MBP has been successfully used by other researchers to generate fusion proteins when the target protein is insoluble and hydrophobic [15,16]. Yet, initial attempts to efficiently express the fusion protein in soluble form met very limited success. Therefore, a series of conditions were optimized, including IPTG concentration and addition of glucose to enhance protein expression level, as well as sonication time to increase the portion of soluble fusion protein. The results suggested that 0.2 mM IPTG and 1% glucose led to a high level expression of the soluble fusion protein. When the combination of optimized conditions was established, the fusion protein was produced with increased solubility. Further, the MBP tag facilitated the purification of the recombinant protein with an MBPTrap HP column, which was specifically designed for the purification of MBP fusion proteins.
To the best of our knowledge, there have been considerable hurdles in attempt to efficiently express the recombinant vp13 due to the hydrophobic and insoluble nature of this protein. The creation of the plasmid encoding MBP-vp13 fusion protein and the development of a process to express the fusion protein as well as the convenient purification method should assist further study of the biology of vp13. The optimized conditions in this study can also be a good reference for others to express and purify proteins that have similar difficulty in soluble expression.
The availability of pure vp13 fusion protein can facilitate the protein’s structural analysis, which in turn can help delineate its biological function. For example, a larger quantity of pure MBP-vp13 could be used for protein crystallization. Protein crystallization has played a vital role in understanding structure and function relationship for many proteins. Hopefully, the materials generated and the process developed in this study will accelerate the understanding of vp13 in its roles in viral replication and pathogenesis. Further understanding the structure and function of vp13 may also facilitate anti-HEV drug development.
More importantly, the methods used in this study to optimize the expression and solubilization of vp13 can be extrapolated to other studies on hydrophobic proteins. Expression and purification of such proteins for vaccines or other immunological reagents can be optimized similarly to enhance the portion of soluble proteins. The soluble proteins are always welcomed due to their proper conformation and function similar to native counterparts.
Acknowledgements
We thank Dr. Suzanne Emerson in NIH and Dr. X-J Meng at Virginia Tech University for some plasmid and antibody used in this study. This study was partially supported by University of Maryland.References
- Kamar N, Dalton HR, Abravanel F, Izopet J (2014) Hepatitis E virus infection. Clin Microbiol Rev 27: 116-138
- Aggarwal R (2010) The global prevalence of hepatitis E virus infection and susceptibility: a systemic review. World Health Organization.
- Geng Y, Yang J, Huang W, Harrison TJ, Zhou Y, et al. (2013) Virus host protein interaction network analysis reveals that the HEV ORF3 protein may interrupt the blood coagulation process. PLoS One 8: e56320.
- Emerson S, Anderson D, Arankalle VA, Meng XJ, Purdy M, Schlauder GG, Tsarev S, (2004) Hepevirus, in: Fauquest CM, Mayo MA, Maniloff J, Desselberger U, Ball LA (Editors) Virus Taxonomy, VIIIth report of the ICTV. Elseiver/Academic Press, London, UK.
- Ahmad I, Holla RP, Jameel S (2011) Molecular virology of hepatitis E virus. Virus Res 161: 47-58.
- Graff J, Nguyen H, Yu C, Elkins WR, St Claire M, et al. (2005) The open reading frame 3 gene of hepatitis E virus contains a cis-reactive element and encodes a protein required for infection of macaques. J Virol 79: 6680-6689.
- Tam AW, Smith MM, Guerra ME, Huang CC, Bradley DW, et al. (1991) Hepatitis E virus (HEV): molecular cloning and sequencing of the full-length viral genome. Virology 185: 120-131.
- Huang YW, Opriessnig T, Halbur PG, Meng XJ (2007) Initiation at the third in-frame AUG codon of open reading frame 3 of the hepatitis E virus is essential for viral infectivity in vivo. J Virol 81: 3018-3026.
- Emerson SU, Nguyen H, Torian U, Purcell RH (2006) ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J Virol 80: 10457-10464.
- Geng Y, Yang J, Huang W, Harrison TJ, et al. (2013) Virus host protein interaction network analysis reveals that the HEV ORF3 protein may interrupt the blood coagulation process. PLoS One 8: e56320.
- Kannan H, Fan S, Patel D, Bossis I, Zhang YJ (2009) The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J Virol 83: 6375-6382.
- Emerson SU, Zhang M, Meng XJ, Nguyen H, St Claire M, et al. (2001) Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc Natl Acad Sci U S A 98: 15270-15275.
- Lee VT, Matewish JM, Kessler JL, Hyodo M, Hayakawa Y, et al. (2007) A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol 65: 1474-1484.
- EMBL (2009) Protein Expression and Purification Core FacilityProtein Expression E.coli. European Molecular Biology Laboratory.
- Hewitt SN, Choi R, Kelley A, Crowther GJ, Napuli AJ, et al. (2011) Expression of proteins in Escherichia coli as fusions with maltose-binding protein to rescue non-expressed targets in a high-throughput protein-expression and purification pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 1006-1009.
- Sorensen HP, Mortensen KK (2005) Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb cell Fact 4: 1.