
Journal of Veterinary Science & Medicine
Download PDF
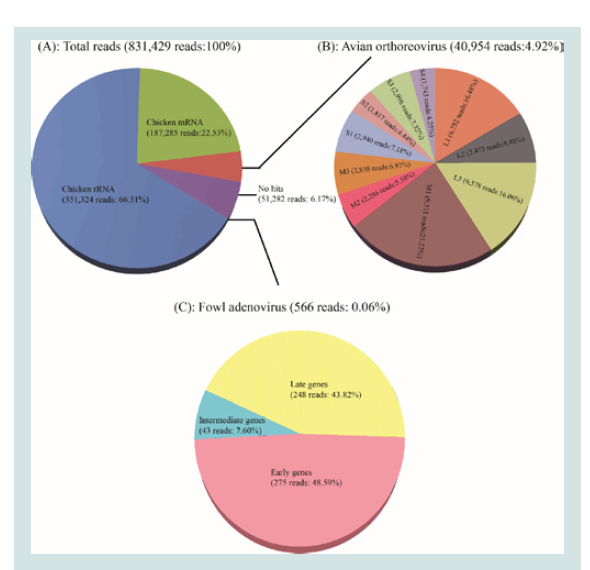 Figure 1: Illustrations of the homology search results for NGS reads and the
sequencing coverage analysis. (A): Total NGS reads homology search result;
(B): The mapping reads of 10 segments of Reo/PA/Layer/27614/13; (C): The
mapping reads to the hexon gene of FAdV/PA/Layer/27614/13.
Figure 1: Illustrations of the homology search results for NGS reads and the
sequencing coverage analysis. (A): Total NGS reads homology search result;
(B): The mapping reads of 10 segments of Reo/PA/Layer/27614/13; (C): The
mapping reads to the hexon gene of FAdV/PA/Layer/27614/13.
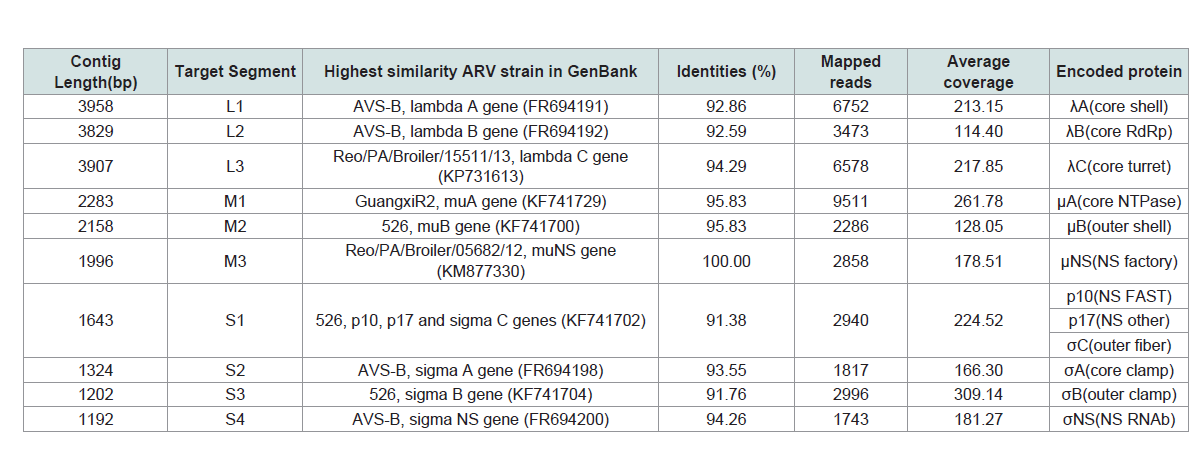 Table 1: De novo assembly and general genome features of the layer-origin avian orthoreovirus (ARV) strain (Reo/PA/Layer/27614/13).
Table 1: De novo assembly and general genome features of the layer-origin avian orthoreovirus (ARV) strain (Reo/PA/Layer/27614/13).
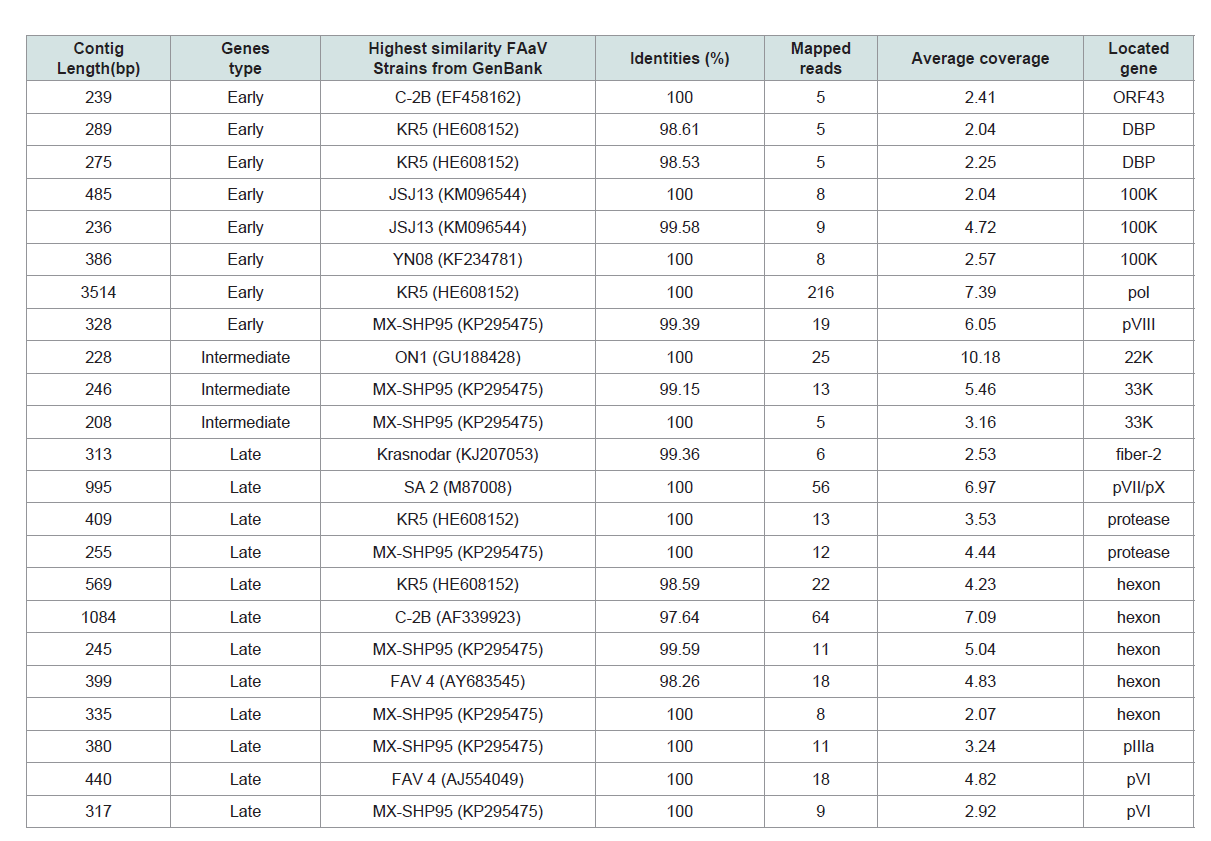 Table 2: De novo assembly of the layer-origin fowl adenovirus (FAdV) strains (FAdV/PA/Layer/27614/13) Complete genome of ARV and partial FAdV transcriptome.
Table 2: De novo assembly of the layer-origin fowl adenovirus (FAdV) strains (FAdV/PA/Layer/27614/13) Complete genome of ARV and partial FAdV transcriptome.
 Figure 2: Phylogenetic analyses. (A): Phylogenetic trees of the μB, σB and
σC genes of Reo/PA/Layer/27614/13; (B): The phylogenetic trees of the
loop1 region on hexon gene of FAdV/PA/Layer/27614/13. Note: The dotmarker and highlighted blue were marked for the two co-infection strains of
Reo/PA/Layer/27614/13 and FAdV/PA/Layer/27614/13 in the phylogenetic
trees.
Figure 2: Phylogenetic analyses. (A): Phylogenetic trees of the μB, σB and
σC genes of Reo/PA/Layer/27614/13; (B): The phylogenetic trees of the
loop1 region on hexon gene of FAdV/PA/Layer/27614/13. Note: The dotmarker and highlighted blue were marked for the two co-infection strains of
Reo/PA/Layer/27614/13 and FAdV/PA/Layer/27614/13 in the phylogenetic
trees.
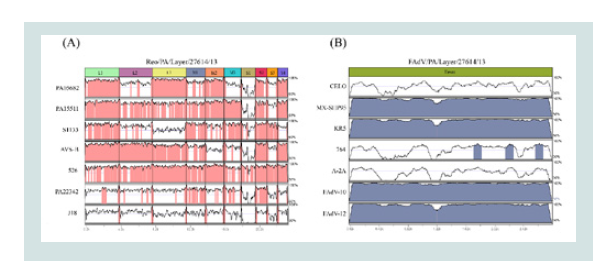 Figure 3: The mVISTA method for whole genome or gene nucleotide alignment.
(A) Alignment result of the Reo/PA/Layer/27614/13 in comparisons with the
Reo/PA/Broiler/05682/12(PA05682), Reo/PA/Broiler/15511/13(PA15511),
S1133, AVS-B,526, Reo/PA/Turkey/22342/13(PA22342) and J18 strains
was illustrated; (B) Alignment result of the FAdV/PA/Layer/27614/13 in
comparisons with the 7 reference strains of the CELO, MX-SHP95, KR5,
764, A-2A, FAaV10 and FAaV12. Note: (1) Areas mapped in pink (A) and
blue (B) represent ≥ 90% similarities; (2) Areas unmapped in white represent
< 90% similarities; (3) The scale bar measures approximate length of the
concatenated genome.
Figure 3: The mVISTA method for whole genome or gene nucleotide alignment.
(A) Alignment result of the Reo/PA/Layer/27614/13 in comparisons with the
Reo/PA/Broiler/05682/12(PA05682), Reo/PA/Broiler/15511/13(PA15511),
S1133, AVS-B,526, Reo/PA/Turkey/22342/13(PA22342) and J18 strains
was illustrated; (B) Alignment result of the FAdV/PA/Layer/27614/13 in
comparisons with the 7 reference strains of the CELO, MX-SHP95, KR5,
764, A-2A, FAaV10 and FAaV12. Note: (1) Areas mapped in pink (A) and
blue (B) represent ≥ 90% similarities; (2) Areas unmapped in white represent
< 90% similarities; (3) The scale bar measures approximate length of the
concatenated genome.
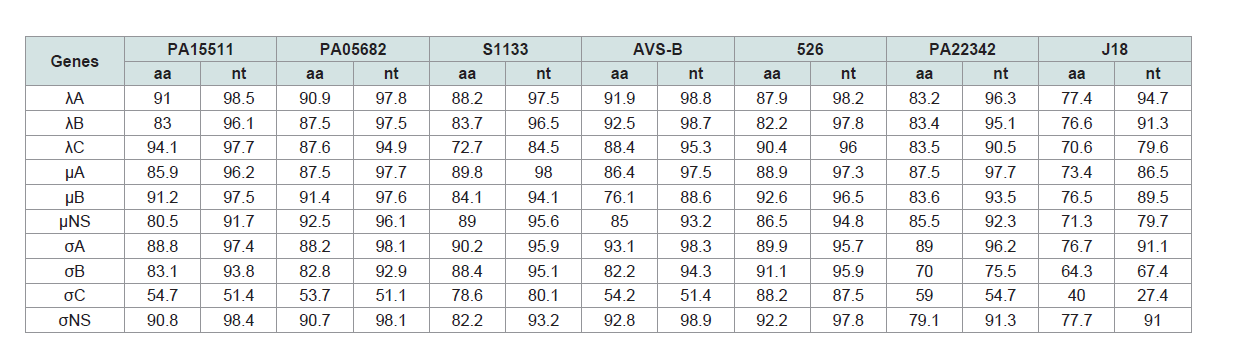 Table 3: Sequence identities of genome segments between the Reo/PA/Layer/27614/13 (Reo/PA27614) strain and orthoreoviruses.
Table 3: Sequence identities of genome segments between the Reo/PA/Layer/27614/13 (Reo/PA27614) strain and orthoreoviruses.
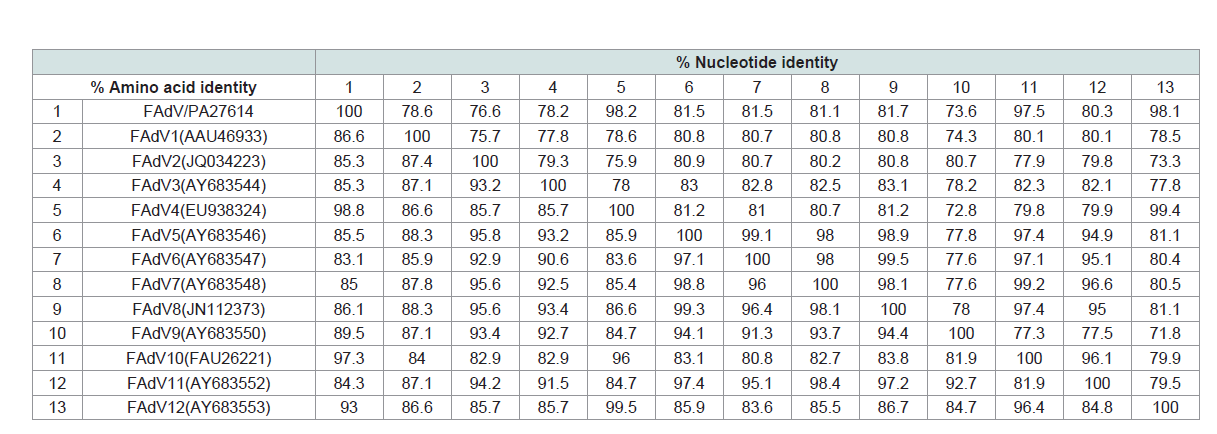 Table 4: Sequence identities of L1 loop of hexon gene between the FAdV/PA/Layer/27614/13 (FAdV/PA27614) strain and fowl adenoviruses.
Table 4: Sequence identities of L1 loop of hexon gene between the FAdV/PA/Layer/27614/13 (FAdV/PA27614) strain and fowl adenoviruses.
Research Article
RNA Deep-Sequencing Analyses for Detection and Characterization of Avian Orthoreovirus and Fowl Adenovirus Co-Infections in Layer Chickens
Tang Y1, Lu H2*
1College of Animal Science and Veterinary Medicine, Shandong
Agricultural University, China
2Department of Veterinary and Biomedical Sciences, Pennsylvania
State University, United States
*Address for Correspondence
Lu H, Wiley Lab/Avian Virology, Department of Veterinary and Biomedical Sciences, The Pennsylvania State University, University
Park, PA 16802, United States, Tel: +1 814 863 4369; Fax: +1 814
865 4717; E-mail address: hxl15@psu.edu
Submission: 19-October, 2019
Accepted: 15-November, 2019
Published: 18-November, 2019
Copyright: © 2019 Tang Y, et al. This is an open access article
distributed under the Creative Commons Attribution License, which
permits unrestricted use, distribution, and reproduction in any medium,
provided the original work is properly cited.
Abstract
Avian orthoreovirus (ARV) and Fowl Adenovirus (FAdV) infections
are pervasive in domestic poultry species, especially in chickens. Coinfections of the two viral pathogens could cause much severer symptoms
on infected birds. In our recent research studies on application of NextGeneration Sequencing (NGS) techniques, we have identified two coinfection viruses of ARV (Reo/PA/Layer/27614/13 or Reo/PA27614) and
FAdV (FAdV/PA/Layer/27614/13 or FAdV/PA27614) from one isolation
from tendon tissue of 35-week-old commercial layer chickens. Among
a total of 831,429 RNA-seq reads, 40,954 reads (4.92%) were confirmed
to be ARV genome sequence, whereas an extremely small number of
566 reads (0.06%) were confirmed to be FAdV mRNA which transcribed
by viral genome DNA. The de novo assembly of two types of viral reads
generated 10 ARV contigs and 23 FAdV contigs, which according to 10
genome segments of ARV full genome and 14 mRNAs of partial FAdV
transcriptome, respectively. Sequence comparison of nucleotide (nt)
and amino acid (aa) sequences of Reo/PA27614 genome and FAdV/
PA27614 hexon gene revealed that the Reo/PA27614 field variant had
40.0-94.1% nt and 27.4-98.8% aa identities in comparison with ARV
reference strains, and the FAdV/PA27614 variant had 73.6-98.2% nt
and 83.1-98.8% aa identities in comparison with FAdV reference strains.
Genome alignment and phylogenetic analysis revealed that the Reo/
PA27614 evolved distant from most ARV reference strains in three major
outer capsid proteins, whereas the FAdV/PA27614 showed a close
relationship with pathogenic reference strains of FAdV group C. Taken
together, the NGS-based deep RNA sequencing techniques allowed us
to identify the RNA virus and DNA virus co-infections at the same time
and provided important epidemiological insights into ARV and FAdV
co-infections in chickens.
Keywords
Avian orthoreovirus; Fowl adenovirus; Layer chicken; Coinfection; Genome; Next-generation sequencing (NGS)
Introduction
As a segmented double-stranded RNA (dsRNA) virus, avian
orthoreovirus (ARV) is the important species in the Orthoreovirus,
one of the 11 genera in the Reoviridae family [1-4]. The full genome
of ARV is comprised by 10 dsRNA segments which are clustered
into three major groups according to mobility in polyacrylamide gel
electrophoresis, namely, large segments (L1,L2, and L3), medium
segments (M1,M2, and M3), and small segments (S1,S2,S3, and S4) [5-7]. Each genome segment of ARV is not used directly for viral protein
synthesis, but is transcribed to form functional mRNA which is
identical to the positive strand of dsRNA [5]. The expression products
of ARV mRNA are 8 structural proteins (λA,λB,λC,μA,μB,σA,σBand
σC) and 4 nonstructural proteins (μNS,p10,p17 and σNS) [4]. Three
of them are major outer capsid proteins (μB,σB and σC) associated
with host cell attachment and induction of virus-neutralizing antibodies [9-12]. The transcription and translation ARV mRNA are
occurred in the cytoplasm of infected cells and the mature virion is
70-80 nm in size without the lipid envelope [13]. ARVs are usually
associated with a variety of clinical diseases in poultry but the viral
arthritis/tenosynovitis, enteric disease, and immunosuppression have
been considered as the primary [14-16].
Members of genus Adenoviridae are medium-sized (90-100
nm), non-enveloped viruses with an icosahedral nucleocapsid
containing a double stranded DNA genomes, which belong to the
Adenoviridae family [17,18]. Based on the isolated species and the
serological differences, avian or fowl adenoviruses (FAdVs) are
currently divided into three groups including conventional FAdV of
group I; Haemorrhagic Enteritis Virus (HEV) and Avian Adenovirus
Splenomegaly Virus (AASV) of group II; and Egg Drop Syndrome
Virus (EDSV) of group III [19,20]. Although chickens are susceptive
to all of the three group viruses, but group I FAdV infections occur
most commonly in commercial chickens worldwide [21]. The group
I FAdVs are sub typed into 12 serotypes in five different subgroups
(A-E) [22]. Because of the great diversities among the 12 serotypes,
different clinical symptoms and pathological lesions associated
with FAdV infections are often observed, including Inclusion
Body Hepatitis (IBH), hydropericardium disease, proventriculitis,
tracheitis and pneumonias [21,23].
Experimental co-infections of ARV and FAdV were reported
in specific-pathogen-free Leghorn chickens for evaluation studies
of gastrointestinal and arthrotropic activity by these two pathogens
[24,25]. However, there was no report for genomic characterization
studies on the ARV and FAdV co-infections naturally occurred in field
chickens. From 2011 to present, the newly emerging ARV variants
have become a major problem in causing severe lameness and arthritis
diseases in Pennsylvania (PA) poultry [26-28]. Additionally, as one of
the most common avian viral disease pathogens, FAdVs were isolated
periodically from our diagnostic broiler and layer cases which were
clinically suspicious to ARV infections. Considering the highly
contagious and pathogenic features of ARV and FAdV in poultry,
their co-infections can cause much severer clinical diseases as our
observations of clinical symptoms during ARV outbreaks occurred in PA in recent years. However, simultaneous virus isolations for both
ARV and FAdV in co-infections of field cases is not easy, traditionally
or commonly, only one type virus (ARV or FAdV) can be isolated or
detected, which could be due to the difference of nucleotide (nt) types
and viral growth kinetics in cell cultures or chicken embryo [29,30].
By using the most advanced Next Generation Sequencing (NGS)
technologies, it has become available to generate large amounts of
sequence data of any virus genome sequences and thus to discover
co-infections of RNA and DNA viruses by RNA deep-sequencing
of the viral genome and transcriptome at the same time [31-33]. In
the present study, we describe our NGS genomic characterization
studies for detection of ARV and FAdV variant co-infections on
one viral isolation made from tendon tissue of field layer chickens,
which provide detail genomic data for the confirmation of naturally
occurring co-infections of ARV and FAdV strains in layer chickens.
Materials and Methods
Virus and virus isolation:
Isolations of various avian viruses from clinical specimens of
diagnostic avian species are routinely conducted at our laboratory.
The diagnostic isolation of ARV field variant strain (Reo/PA/
Layer/27614/13, or Reo/PA27614) used in this study was isolated
from tendon tissue of 35 weeks-old layer chickens from a flock
experienced feather loss and egg production drop. The ARV isolation
and identification tests were conducted per procedures described in
our previous publications [26-28]. Briefly, 1) tendon tissue of the layer
chickens showed symptoms of ARV infections was processed for
virus isolation in LMH cell (CRL-2113, ATCC) cultures for 2-3 serial
cell passages; 2) ARV-infected LMH cells, which were characterized
by “bloom-like” giant Cytopathic Effect (CPE) cells, were harvested
and prepared on a glass slide for ARV identification test; and 3) ARV
positive isolates were confirmed by ARV Fluorescent Antibody (FA)
(Ref No. 680, VDL 9501, NVSL, Ames, IA) staining the ARV-infected
CPE cells.
RT-PCR and σC gene sequencing of ARV:
Total RNA was extracted from the ARV isolate (Reo/PA27614)
using an RNeasy Mini Kit (Cat. No. Z74106, QIAGEN, Valencia, CA,
USA). The RT-PCR amplification of σC gene was carried out using
P1 and P4 primers with a One Step RT-PCR Kit (Cat. No.210212,
QIAGEN, Valencia, CA, USA) [34]. The RT-PCR products, obtained
through 1% agarose gel electrophoresis, were purified using a gel
extraction kit (Cat. No.04113KE1, Axygen, Tewksbury, MA, USA)
per the manufacturer’s protocol and then were directly submitted
to Penn State Genomics Core Facility at University Park campus for
Sanger sequencing.
PCR amplification of FAdV hexon gene from the ARV isolate:
Total DNA from the ARV isolate (Reo/PA27614) described
above was extracted using a DNeasy Blood & Tissue Kit (Cat. No.
69504, QIAGEN, Valencia, CA, USA) according to manufacturer
instruction. The PCR amplification of hexon gene was carried out
using H1 and H2 primers with a Taq PCR Master Mix Kit (Cat. No.
201443, QIAGEN, Valencia, CA, USA) [35]. The PCR products were
isolated through electrophoresis using 1% agarose gel with ethidium
bromide and visualized under UV light, and then were purified and submitted to Penn State Genomics Core Facility at University Park
campus for Sanger sequencing.
Next-generation sequencing:
RNA libraries were constructed from 1 μg of DNase-treated
total RNA samples using the TruSeq Stranded Total RNA Sample
Prep Kit (Cat. No. RS-122-2201, Illumina, San Diego, CA, USA)
according the manufacturer’s protocol but without the initial poly-A
enrichment step. Briefly, the total RNA was fragmented into small
pieces using 5x fragmentation buffer under elevated temperature
[36]. First strand cDNA was synthesized using random hexamer
primer and SuperScript®
II reverse transcriptase (Cat. No. 18064-014,
Invitrogen, Grand Island, NY, USA). The second-strand cDNA was
synthesized using RNase H (Cat. No. 18021-071, Invitrogen, Grand
Island, NY, USA) and DNA polymerase I (Cat. No. M0209S, New
England BioLabs, Ipswich, MA, USA). The double-stranded cDNA
was purified by a QIAquick PCR extraction kit (Cat. No. 28104,
Invitrogen, Grand Island, NY, USA), and end repair were performed
before the ligation of sequencing adaptors. The library size and
quality were checked by Agilent Bioanalyzer (Agilent Technologies,
Santa Clara, CA, USA). The library product was directly sequenced
via Illumina MiSeq using 150-nt single-read sequencing according to
the manufacturer’s protocol.
De novo assembly of viral genome:
De novo assembly and analyzing of NGS raw data were carried
out by different modules in “NGS Core Tools” and “De Novo
Sequencing” main tools of CLC Genomics Workbench V7.5.2
software (QIAGEN, Boston, MA, USA). Briefly, sequencing adaptors,
reads mapping to chicken rRNA or mRNA reads, and low-quality
reads were trimmed off by “Trim Sequences” module before further
processing. The clean reads were assessed through “De Novo
Assembly” module to get assembled contiguous sequences (contigs).
To identify the origin of the assembled contigs, the sequence of the
contigs were extracted and submitted to “BLAST at NCBI” module.
Based on the BLASTN searching results, all ARV-homologous and
FAdV homologous contigs were selected as target sequences to build
the full-genome of ARV and the transcriptome of FAdV. By remapping the NGS raw reads to the viral contigs of two viruses using “Map Reads to Reference” module, the target contigs were further
improved in length and sequencing coverage. Finally, the consensus
sequences were obtained and considered as the final assembly of ARV
genome and FAdV transcriptome.
Sequence analyses:
To predict the viral Open Reading Frames (ORFs), align the
homologous segments of genes, and identify the sequence similarities
in pairwise, the tools of EditSeq and MegAlign of DNASTAR
Lasergene 12 Core Suite (DNASTAR, Inc. Madison, WI, USA) were
employed. The highest sequence identities searching in Genbank
were carried out by BLASTN online program (http://blast.ncbi.nlm.
nih.gov/Blast.cgi). Sequencing coverage and reads mapping of each
assembled viral contigs were calculated by “Map Reads to Reference”
module of CLC Genomic Workbench V7.5.2 software (QIAGEN,
Boston, MA, USA) and visualized in pie chart using PowerPoint 2010
(Microsoft, Redmond, WA, USA). Phylogenetic trees of viral genes
were estimated in MEGA 6 program using a neighbor-joining model
and the bootstrap validation method with 1000 replications [37].
Visualized whole-genome or gene alignment results were generated
by mVISTA online program (http://genome.lbl.gov/vista/mvista/
submit.shtml) and the studied ARV genome and FAdV hexon gene
were set as scales.
Genbank accession numbers:
The ARV full genome sequence and FAdV hexon gene sequence
obtained in this study have been deposited in the Genbank under
the accession numbers of KU169288 - KU169297 and KT428298.
The ARV reference strains Reo/PA/Broiler/05682/12 (or PA05682),
Reo/PA/Broiler/15511/13 (or PA15511), S1133, AVS-B, 526,
Reo/PA/Turkey/22342/13 (or PA22342) and J18 were listed in
(Supplementary Table S1). The full-length of hexon gene sequences
of FAdV reference strains, CELO, MX-SHP95,
Results
RT-PCR, PCR and Sanger sequencing:
The S1-based one-step RT-PCR using P1/P4 primers successfully
amplified viral RNA of the ARV field variant strain (Reo/PA27614) at
the 1088bp position. Sanger sequencing results of the ARV variant’s
PCR product (KP727769) revealed about 91% nt identities with the
most similarity ARV strain in GenBank (KF741702). Unfortunately,
our attempt to obtain the FAdV hexon gene was not successful in
amplifying the estimate 1219bp PCR product.
NGS raw data processing:
After removing low-quality reads and trimming sequencing
adapter through the Quality Control (QC) filters of the Illumina
Miseq sequencer, a total of 831,429 sequencing reads were outputted
in a 238Mb fast q format file. By using BLASTN searching, the reads
mapped to the mRNA and rRNA of chicken or other origins were
finally confirmed and considered as the contamination or non-target
reads. As a result among the 831,429 reads, 551,324 reads (66.31%)
were identified to be the chicken rRNA source and 187,285 reads
(22.53%) to be the chicken mRNA source (Figure 1A). The remaining
92,792 reads (11.16%) were identified as the clean reads that consisted
of ARV genome group (40,954 reads, 4.92%), no hits group (51,282
reads, 6.17%), and FAdV transcriptome group (566 reads, 0.06%)
(Figure 1A).
De novo assembly:
The total of 92,792 clean reads described above were subject to
de novo assembly of viral contigs. After processing through the “De
Novo Assembly” module of CLC Genomics Workbench software, a
total of 131 contigs were generated with length from 50nt to 3958nt.
The mapped reads of assembled contigs were calculated at various
numbers (2 reads to 613 reads), which resulted the average coverage
ranging from 2.04x to 309x. BLASTN online searching results revealed
the existing ten ARV associated contigs with length from 1192nt to
3958nt and 23 FAdV associated contigs with length from 208nt to
3514nt among total assembled contigs (Table 1 and Table 2). After the most
homology sequence searching in Genbank, ten ARV contigs showed
high nt similarities (91.8%-100%) with the published strains and
FAdV contigs showed higher nt similarities (97.64%-100%) with the
published strains (Table 1 and 2). The initial alignment of ARV and
FAdV contigs with the most homology reference sequences indicated
that the size of the ARV contigs exactly matched the full-length of
10 ARV genome segments, respectively, whereas most FAdV contigs
were only partial sequences of the target mRNAs.
By mapping back NGS raw reads to the 10 ARV contigs, the length
and sequencing coverage of assembled contigs were further improved
to yield the consensus sequences as final ARV genome segments. The
mapped reads of each segment were summarized in (Table 1 and
Figure 1B). The full-genome of this ARV variant, Reo/PA27614, was
23,495 bp in size contained 10 genome segments ranged from 1192
bp (S4) to 3958 bp (L1). Nine out of ten segments are monocistronic
and only S1 segment is tricistronic. These segments encoded 12 viral
proteins and the length of ORFs ranged from 297 bp (p10) to 3882
bp (λA), which were identical to published strains of these general
ARV features. At the 5’ and 3’ termini of the each genome segment of
the Reo/PA27614 variant strain located between 12bp to 98bp of the
Untranslated Regions (UTRs). By aligning the 5’ UTRs and 3’ UTRs,
respectively, the highly conserved terminal sequence was confirmed
at 5’ UTR (5’-GCUUUU-3’) and 3’ UTR (5’-UCAUC-3’). These
common genome features of the Reo/PA27614 variant strain were
similar to other published ARV reference strains.
By using the same contig improvement method as ARV, the
partial transcriptome and mapped reads of FAdV strain, FAdV/
PA27614, were finally obtained (Table 2 and Figure 1C). The resulting
FAdV/PA27614 mRNA clusters were divided into three main groups.
Group 1 was early genes group included 8 contigs corresponding to 5
viral genes that were expressed immediately after the infection. Group 2 was intermediate gene group included only 3 contigs corresponding
to 2 viral genes (22K and 33K) that located proximal to the terminus
of the upper and lower strands of the genome. The late gene group
(group 3) was the largest group included 12 contigs corresponding
to 6 viral that mainly located in the central region of the genome.
Five out of twelve contigs is targeted on the most important FAdV
structural protein of hexon. By further aligning five hexon gene
contigs to reference sequence (KP295475), the full hexon gene of the
FAdV/PA27614 strain was successfully assembled at length of 2,814
bp.
Sequence comparisons:
The nucleotide (nt) and amino acid (aa) of ten genes of Reo/
PA27614 strain were compared in their homologs with seven
ARV strains, including two PA broiler ARV strains (PA15511 and
PA05682), three broiler ARV reference strains (S1133, AVS-B and
526), one PA turkey ARV strain (PA22342), and one duck ARV
reference strain (J18) (Supplementary Table S1). The pairwise nt and
aa comparisons between the Reo/PA27614 strain and chicken origin
ARV strains revealed low to high similarities (nt: 53.7-94.1%; aa: 51.1-
98.8%), which were higher than those of turkey origin strains PA22342
(nt: 59.0-89.0%; aa: 54.7-97.7%) and duck origin strains J18 (nt: 40.0-
77.7%; aa: 27.4-94.7%). The highest identities for individual genes
between the Reo/PA27614 strain and reference strains were showing
heterogeneities (Table 3), meaning that the most homologous strain
for each gene is different. In general, the AVS-B strain was considered
as owning the largest number (total 4 genes) of highest identity (nt:
91.9-92.8%; aa: 98.3-98.9%) segments with Reo/PA27614 including
the in λA-, λB-, σA- and σNS-encoding genes among all compared
strains. For other six genes, the Reo/PA27614 strain showed highest
identity with the 526 strain in μB-, σB- and σC-encoding genes (nt:
88.2-92.6%; aa: 87.5-96.5%), the PA15511 in λC-encoding gene (nt:
94.1%; aa: 97.7%), the PA05682 strain in μNS-encoding gene (nt:
92.5%; aa: 96.1%), and the S1133 strain in μA-encoding gene (nt:
89.8%; aa: 98.0%).
The pairwise nt and aa comparisons of the loop 1 region (residues
101 to 298) on hexon gene were carried out between the FAdV/
PA27614 strain and 12 serotypes of FAdV reference strains (Table 3). Overall, the FAdV/PA27614 strain showed highest identity with
the FAdV-4 in the homologous gene region (nt: 98.2%; aa: 98.8%)
belonging to subgroup C of FAdVs. For the nt sequence comparison
results between FAdV/PA27614 and other 11 FAdV serotypes, the
relatively high identities were observed in FAdV10 and FAdV12 (nt:
>97.5%), but lower in compared with FAdV1, FAdV2, FAdV3 and
FAdV9 (nt: <81.7%). When compared sequence similarities in aa
with non-FAdV4 strains, FAdV/PA27614 showed high similarities
with FAdV9, FAdV10 and FAdV12 (aa: >89.5%) which belonged to
the same group as FAdV4 (subgroup C). Interestingly, as the typically
representative serotype of FAdV subgroup A, FAdV1 also showed
relatively high identity with FAdV/PA27614 (aa: 86.6%), indicating
the next closest relationship of the studied strain to subgroup A of
FAdVs. However, the other 7 serotypes of FAdVs form subgroup B, D
and E showed lower identities with FAdV/PA27614 (aa: <86.1%) and
the lowest identity (aa: 83.1%) were found between FAdV/PA27614
and FAdV6, which belonged to the subgroup D of FAdVs.
Phylogenetic analysis of the Reo/PA27614 and FAdV/PA27614:
To study the evolutionary relationships of the Reo/PA27614 strain
with other ARV reference strains, the nt sequence of three major
outer capsid encoding genes proteins (μB, σB and σC) were subjected
to phylogenetic-tree analysis using rooted maximum likelihood
method (Figure 2A, μB). For μB gene analysis, four genotyping
lineages were formed by the Reo/PA27614 strain and reference
strains and no specific host-associated relationships were identified
between these lineages. The Reo/PA27614 strain together with two
PA broiler ARV field strains (PA05682 and P15511) and one classic
ARV strain 138, formed the lineage II group, and the studied strain
showed closer relationship with two PA field strains than 138 strain.
In contrast with μB gene, the phylogenetic tree of σB gene revealed
four host-associated groups which formed by Reo/PA27614 and
reference strains (Figure 2A, σB). Although Reo/PA27614 strain was
located at chicken I group with most classic ARV reference strains,
but only showing distant relatedness. As the most diverse gene of
ARV, σC phylogenetic analysis using the Reo/PA27614 strain and
reference strains generated five genotyping clusters which showing
less than 70% nt identity between any two clusters (Figure 2A, σC).
The Reo/PA27614 was classified as a member of cluster 1 (PA01224a),
exhibiting significant divergence with most included strains, even the
reference strains in the same cluster, which confirmed the sequence
comparison results as described above.
The evolutionary relationships between the FAdV/PA27614
strain and other FAdVs were shown in (Figure 2B). All analyzed
FAdV strains were clustered into five major groups (A-E). Although
the FAdV/PA27614 was clustered into the C group with the FAdV reference strains isolated in different countries, it also closely related
to two FAdV1 strains of A group which consistent with pairwise
comparison results as described above.
The visualized genome or gene alignments:
The mVISTA online program aligned whole genomes of Reo/
PA27614 and reference ARV strains and visualized the sequence
identities of individual genome segments between them (Figure 3A). The classic ARV reference strain 526 showed a continuous
high genetic relatedness (nt: >90%) with Reo/PA27614 throughout
whole genome. The highest related segments between the study strain
and reference strains were found at L1, L2 segments of AVS-B and
L3 segment of PA05682 with more than 95% nt identities in most
regions of these segments. The turkey-origin PA22342 strain shared
moderate sequence identities with Reo/PA27614 of the study strain
throughout most whole genomes, only M1 and S2 segments showed
higher similarity between them. The duck-origin J18 strain shared low
genetic relatedness with Reo/PA27614 throughout whole genomes,
and an even lower identity was observed in S1 segment (nt: <50%),
only showing high identities in the 5’and 3’ termini of each segment.
The visualized hexon gene alignments of FAdVs revealed wideranging genetic relatedness between FAdV/PA27614 strain and
FAdV4 reference strains (MX-SHP95 and KR5) (Figure 3B). The
FAdV10 and FAdV12 were also showed high identities with FAdV/
PA27614 throughout the whole hexon gene and FAdV10 was
consider as the closest strain to FAdV/PA27614 among all reference
strains. The CELO strain shared moderate sequence identities with
FAdV/PA27614, whereas the 764 and A-2A strains only showed
shared low sequence identities with FAdV/PA27614, especially from
303nt-894nt which corresponding to region of L1 loop (nt: <50%).
Discussion
Many research studies have indicated that ARV-infections in
poultry can cause various clinical symptoms [2,38], particularly
severe viral arthritis or tenosynovitis, runting-stunting syndrome,
enteric disease and malabsorption syndromes [2,32-34]. The newly
emerged/emerging ARV field variant strains have been detected in
various poultry species including broilers, broiler breeders, layers,
turkeys, chukar partridges, guinea fowls, pheasants and quails in PA
during the last several years, and severe viral arthritis or tenosynovitis
are the most common symptoms seen in ARV-affected poultry [26-28,39,41].
In addition to ARV infections, FAdV is another ubiquitous
pathogen in poultry farms and pathogenic FAdV strains may cause
clinical diseases but their pathogenic roles were not well studied
or remained questionable in the past [21]. As published studies
indicated that only FAdV4 was confirmed as a causative agent of
broiler disease called infectious hydropericardium, Angara disease
or hepatitis and Hydropericardium Syndrome (HHS) [42]. The HHS
affected broiler flocks were seen mainly at 3 to 5 weeks of age and
the mortality rate could be up to 75%. Research findings showed that
the precondition of immunosuppression in chickens could lead to an
increased intensity and severity of HHS by synergistic effect under
experimental conditions [43]. In recent years in China, FAdV4 and
FAdV8 have been confirmed the severely pathogenic strains which
caused significant losses in broiler chickens and ducks [44-46]. ARV as an immunosuppressive agent, it could be accompanying initial
infections or secondary infections during FAdV epidemic outbreaks.
Thus in field conditions, ARVs can not only induce primary
tenosynovitis in chickens, but also aggravate symptoms of FAdVassociated HHS.
Genomic characterization finding of the ARV and FAdV strains
in one isolation described in the present study is the first report of
these two viruses’ co-infections naturally occurred and detected in
commercial layer chickens, which provides scientific methodology
and important epidemiological insights for detection of co-infections
and genomic characterization of RNA and DNA viruses from
virus isolations or clinically infected animals. This specific layer
chicken isolate was one of more than 20 other layer and broiler
ARV isolates we obtained from diseased flocks and selected for full
genome sequencing characterization studies. By using pairwise nt
and aa sequence comparisons, we found the AVS-B strain had the
largest number of highest identity segment with the ARV variant
Reo/PA27614 described in this study, and we also found that at
least one highest identity segment existed in each of other reference
ARV strains. Indeed, segments 3 was the most homologous segment
numbers of the ARV 526 strain, indicating the AVS-B and 526
strains may mainly contribute to the origin of Reo/PA27614 variant
by terming reassortment. Each of the PA broiler ARV field strains
of PA05682 and PA15511 also shared most homology L3 and
M3 segments with Reo/PA27614, respectively, indicating further
reassortments may occur between the original reassortant strain and
ARV field strains during infections in poultry.
Sequence homology and phylogenetic analysis of the major outer
capsid proteins (μB,σB and σC) of the newly isolated ARV revealed
that these proteins were originated from ARV 526 strain. As the
important structural proteins, μB was involved in virus entry and
transcriptase activation [47]; σB was responsible for inducing groupspecific neutralizing antibodies [48]; and σC played an important
role for virus attachment and acted as an apoptosis inducer [9,49].
Therefore, the Reo/PA27614 variant strain in present study may
have the same serological and infection features with the ARV 526
strain. In addition, the mVISTA alignment results of ARVs also
revealed that the ARV 526 strain shared continually high sequences
of identities with Reo/PA27614 variant strain throughout the whole
genome, whereas other ARV reference strains only shared high
sequences of identities with the Reo/PA27614 variant strain in some
non-continually segments. In this case, we can further speculate
that there may be a series of reassortments and mutations on ARV
526 strain and lead to the generation or reassortments for the Reo/
PA27614 variant strain, which was the major co-infection virus we
described in this study.
Because the transcriptome of the FAdV/PA27614 strain was
partially identified in the present study, the sequence comparison
and phylogenetic analysis of FAdV/PA27614 were carried out using
only hexon gene which fully obtained through the RNA-seq. The
hexon protein of FAdV is the major capsid protein which contains
type-, group-, and subgroup-specific antigenic determinants [21],
thus most detection, differentiation, comparison and phylogenetic
analysis of FAdVs were conducted based on the most variable region
of loop 1 on hexon [50]. The comparison of aa and nt sequence of the hexon loop 1 region revealed that the FAdV/PA27614 strain
was belonging to FAdV4 serotypes and also shared high identities
with FAdV9, FAdV10 and FAdV12 strains. Phylogenetic analysis
indicated the FAdV/PA27614 together with the FAdV9, FAdV10
and FAdV12 reference strains were clustered into genotype C group.
The members of this group also included most pathogenic strains of
FAdV4 which isolated worldwide in recent years and some of them
associated with HHS. The close relationship between FAdV/PA27614
and FAV4 pathogenic strains was not only showed at loop 1 region,
but also showed at full-length of hexon which confirmed by mVISA
alignment. Base on the above sequence comparison and analysis,
FAdV/PA27614 was likely to be a pathogenic strain which could
cause the HHS in broiler chickens.
In this study, our routine virus isolation tests showed that the
Reo/PA27614 variant caused the significant CPEs of cell fusion on
LMH cells, , whereas the formation of FAdV CPE was not observed
or occurred in this case, which was possibly due to the very low
population of FAdV/PA27614 in the sample and also the dominated
fast growth of the ARV, thus PCR or traditional immunoassays
failed in detection of FAdV/PA27614 in this co-infection case.
Fortunately, the most advanced NGS technology for metagenomics
studies provides a powerful tool for the conduct of a fast and highthroughput sequencing of genomes in a wide range of organisms
from viruses to mammalian genomes [51,52]. By employing a deep
RNA sequencing technique, we successfully identified ARV genome
and FAdV transcriptome from a single isolate. The mapping reads
of ARV genome is 40,954 (4.92% of total reads) which was much
higher than that of FAdV transcriptome 566 (0.06% of total reads),
indicating that there was a huge difference between the amount of
ARV viral RNA and FAdV mRNA in the sequencing sample. Such
difference may associated with the numbers of the viruses in the
original tissue specimen or the viral characteristic of growth kinetics
in LMH cell culture [53]. Although the transcriptome of the FAdV/
PA27614 strain was partial, we made successful in assembling the
full-length of the hexon gene and carrying out the sufficient sequence
analyses for the characterization of the FAdV/PA27614 strain
In summary, we obtained the detailed genomic information of
naturally occurred co-infections of ARV and FAdV variant strains in
one isolation from layer chickens using NGS deep-sequencing analyses,
providing a research methodology for genomic characterizing the coinfections of RNA and DNA viruses. By using the comprehensive
sequence analyses, we identified that the Reo/PA27614 variant strain
was a ressortant virus with its genome segments from both historical
ARV strains and the newly emerged ARV field variant strains; the
FAdV/PA27614 strain was closely related with FAdV4 pathogenic
strain and could be associated with HHS disease. The findings of this
study indicate that one virus isolate could contain both detectable and
undetectable viruses by traditional virus identification tests. Thus,
genomic characterizations provide the most advanced technique in
detecting all viruses by their genome sequences, which is particularly
useful in correct selections of autogenous vaccine candidates from
field virus isolations.
Supplementary Materials:
Author Contributions:Project conductors: T.Y. and H.L.; whole
viral genome sequencing and NGS data analysis: T.Y.; manuscript draft: T.Y.; manuscript review and edit: H.L..
Reference
Acknowledgment
The avian reovirus research projects were funded by The
Pennsylvania Poultry Industry Broiler/Egg Check-Off Research
Program in 2016/2018, and The Pennsylvania Soybean Board
Research Program in 2018, Pennsylvania, USA.
Citation
Tang Y, Lu H. RNA Deep-Sequencing Analyses for Detection and Characterization of Avian Orthoreovirus and Fowl Adenovirus Co-Infections in Layer Chickens. J Veter Sci Med. 2019;7(2): 9.