
Journal of Hematology & Thrombosis
Download PDF
Letter to the Editor
*Address for Correspondence: Nobuyoshi Hanaoka, MD, Ph D, Department of Hematology/Oncology, Wakayama Medical University, 811-1 Kimiidera, Wakayama 641-8510, Japan, Tel: +81 73 4410665; Fax: +81 73 4410653; E-mail: nhanaoka@wakayama-med.ac.jp
Citation: Hanaoka N, Mushino T, Murata S, Nagakura S, Horikawa K, et al. Racial Differences in Paroxysmal Nocturnal Hemoglobinuria Thrombosis and Glycosylphosphatidylinositol-Deficient Granulocytes. J Hematol Thromb 2016;2(2): 2.
Copyright © 2016 Hanaoka et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.Journal of Hematology & Thrombosis | ISSN: 2380-6842 | Volume: 2, Issue: 2
Submission: 05 October, 2016 | Accepted: 17 October, 2016 | Published: 19 October, 2016
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired glycosylphosphatidylinositol (GPI)-deficient hematopoietic stem cell disorder that is characterized by complement-mediated hemolytic anemia, thrombosis, and bone marrow failure. Eculizumab, an inhibitor of terminal complement C5, provides patients with PNH with a good quality of life by ameliorating hemolysis and thrombosis, and promotes a better understanding of the complement-mediated molecular pathophysiology of PNH [1-3]. However, the underlying reasons for distinct differences in the incidences of thrombosis and bone marrow failure between Caucasian and Asian patients with PNH are still unknown (West vs. Japan, 32% vs. 4%) [4]. Racial differences are more evident in PNH thrombosis than in other conventional arterial thromboses (PNH vs. others, 2.4 to 9.8 vs. 1.7 to 3.9-fold increase) [4,5], suggesting that the presence of thrombus formation is characteristic of PNH. Progression to thrombosis is generally attributable to synergistic interactions between hypercoagulation and hypofibrinolysis. Hypercoagulability in PNH patients may be due to chronic hemolysis, accentuated platelet aggregation, and other complement-mediated events, whereas hypofibrinolysis in PNH patients is explained by the loss of the urokinase plasminogen activator receptor (uPAR) on leukocytes and elevated soluble uPAR plasma levels [3,6]. Besides erythrocytes and platelets, leukocytes play a critical role in the progression of thrombosis. Thrombotic events often occur in patients with leukocytosis due to myeloproliferative neoplasms as well as in those treated with granulocyte macrophage colony-stimulating factor. In contrast, patients with a plastic anemia-PNH syndrome showing leukocytopenia manifest clinical thrombosis less frequently than patients with de novo PNH maintaining normal leukocyte counts [7,8]. In contrast to thrombosis, cytopenias are more common in Japanese PNH patients than in those in the United States [4]. Based on this background, we attempted to elucidate a plausible link between marrow failure-associated leukocytopenia and the infrequent complication of thrombosis in Japanese PNH patients by analyzing data obtained from our own PNH patients and previous findings [1-3,6-10]. We also investigated the reasons for racial differences using GPI-deficient (PNH) granulocyte counts in PNH thrombosis.

Racial Differences in Paroxysmal Nocturnal Hemoglobinuria Thrombosis and Glycosylphosphatidylinositol-Deficient Granulocytes
Nobuyoshi Hanaoka1*, Toshiki Mushino1, Shogo Murata1, Shoichi Nagakura2, Kentaro Horikawa3, Tatsuya Kawaguchi4, Takashi Sonoki1 and Hideki Nakakuma1
- 1Department of Hematology/Oncology, Wakayama Medical University, Wakayama, Japan
- 2Department of Hematology, National Hospital Organization Kumamoto Minami Hospital, Kumamoto, Japan
- 3Koshi Dai-Ichi Hospital, Kumamoto, Japan
- 4Departments of Hematology and Infectious Diseases, Kumamoto University Graduate School of Medical Sciences, Japan
*Address for Correspondence: Nobuyoshi Hanaoka, MD, Ph D, Department of Hematology/Oncology, Wakayama Medical University, 811-1 Kimiidera, Wakayama 641-8510, Japan, Tel: +81 73 4410665; Fax: +81 73 4410653; E-mail: nhanaoka@wakayama-med.ac.jp
Citation: Hanaoka N, Mushino T, Murata S, Nagakura S, Horikawa K, et al. Racial Differences in Paroxysmal Nocturnal Hemoglobinuria Thrombosis and Glycosylphosphatidylinositol-Deficient Granulocytes. J Hematol Thromb 2016;2(2): 2.
Copyright © 2016 Hanaoka et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.Journal of Hematology & Thrombosis | ISSN: 2380-6842 | Volume: 2, Issue: 2
Submission: 05 October, 2016 | Accepted: 17 October, 2016 | Published: 19 October, 2016
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired glycosylphosphatidylinositol (GPI)-deficient hematopoietic stem cell disorder that is characterized by complement-mediated hemolytic anemia, thrombosis, and bone marrow failure. Eculizumab, an inhibitor of terminal complement C5, provides patients with PNH with a good quality of life by ameliorating hemolysis and thrombosis, and promotes a better understanding of the complement-mediated molecular pathophysiology of PNH [1-3]. However, the underlying reasons for distinct differences in the incidences of thrombosis and bone marrow failure between Caucasian and Asian patients with PNH are still unknown (West vs. Japan, 32% vs. 4%) [4]. Racial differences are more evident in PNH thrombosis than in other conventional arterial thromboses (PNH vs. others, 2.4 to 9.8 vs. 1.7 to 3.9-fold increase) [4,5], suggesting that the presence of thrombus formation is characteristic of PNH. Progression to thrombosis is generally attributable to synergistic interactions between hypercoagulation and hypofibrinolysis. Hypercoagulability in PNH patients may be due to chronic hemolysis, accentuated platelet aggregation, and other complement-mediated events, whereas hypofibrinolysis in PNH patients is explained by the loss of the urokinase plasminogen activator receptor (uPAR) on leukocytes and elevated soluble uPAR plasma levels [3,6]. Besides erythrocytes and platelets, leukocytes play a critical role in the progression of thrombosis. Thrombotic events often occur in patients with leukocytosis due to myeloproliferative neoplasms as well as in those treated with granulocyte macrophage colony-stimulating factor. In contrast, patients with a plastic anemia-PNH syndrome showing leukocytopenia manifest clinical thrombosis less frequently than patients with de novo PNH maintaining normal leukocyte counts [7,8]. In contrast to thrombosis, cytopenias are more common in Japanese PNH patients than in those in the United States [4]. Based on this background, we attempted to elucidate a plausible link between marrow failure-associated leukocytopenia and the infrequent complication of thrombosis in Japanese PNH patients by analyzing data obtained from our own PNH patients and previous findings [1-3,6-10]. We also investigated the reasons for racial differences using GPI-deficient (PNH) granulocyte counts in PNH thrombosis.
Figure 1A shows a wide range of D-dimer levels as an incidence of fibrinolysis in PNH patients with thrombosis. D-dimer levels in 11 Western PNH patients with thrombosis were significantly higher than those in 6 Japanese patients with thrombosis. These results suggest that a hypercoagulable state rather than hypofibrinolysis in PNH thrombosis is more common in Western PNH patients than in Japanese patients. Thrombosis frequently occurred in PNH patients with a higher leukocyte count (mean leukocyte count in patients with thrombotic events (TE+) vs. no TE (TE-), 5.1x108/l vs. 3.9x108/l; p=0.006) and more PNH granulocytes (mean number of PNH granulocytes in patients with TE+ vs. TE-, 4.4x108/l vs. 2.4x108/l; p< 0.001) (Figure 1B). On the other hand, patients with a large percentage of PNH clones, i.e. PNH granulocytes >50%, also showed racial differences in PNH thrombosis, whereas these differences disappeared when the percentage of PNH granulocytes decreased (Figure 1C). These results suggest that thrombus formation in PNH is characterized by a hypercoagulable state attributable to PNH granulocytes coupled with PNH hemolysis. Of note, the racial differences disappeared when assembling PNH patients with a total leukocyte count ≥3.0x108/l among patients showing evident hemolysis with more than 50% PNH granulocytes (Figure 1D). Taken together, although hemolysis contributes to the incidence of PNH thrombosis (data not shown), which supports the allegedly identical amelioration of PNH thrombosis after the administration of eculizumab [1,3], it is no longer regarded as a major cause of racial differences (Figure 1C). Of interest, racial differences were detected among patients with a total leukocyte count < 3.0x108/l containing more than 50% PNH clones (Figure 1D), which implies that underlying factors such as genetic conditions are involved in conventional thrombus formation, even in PNH patients, because these factors make PNH thrombosis likely to be less affected by a possible specific feature of PNH granulocytes and hemolysis. Alternatively, the number of platelets was not a factor causing racial differences (Figure 1E), indicating that simple bone marrow failure could not allow conferring a biological distinction of PNH thrombosis contributing to racial differences. Therefore, we attribute racial differences in PNH thrombosis to the epifunctional causes of PNH granulocytes associated with hypercoagulation, including that through a potential molecule specifically expressed or absent on PNH granulocytes. However, larger studies are needed in order to reach more concrete conclusions.
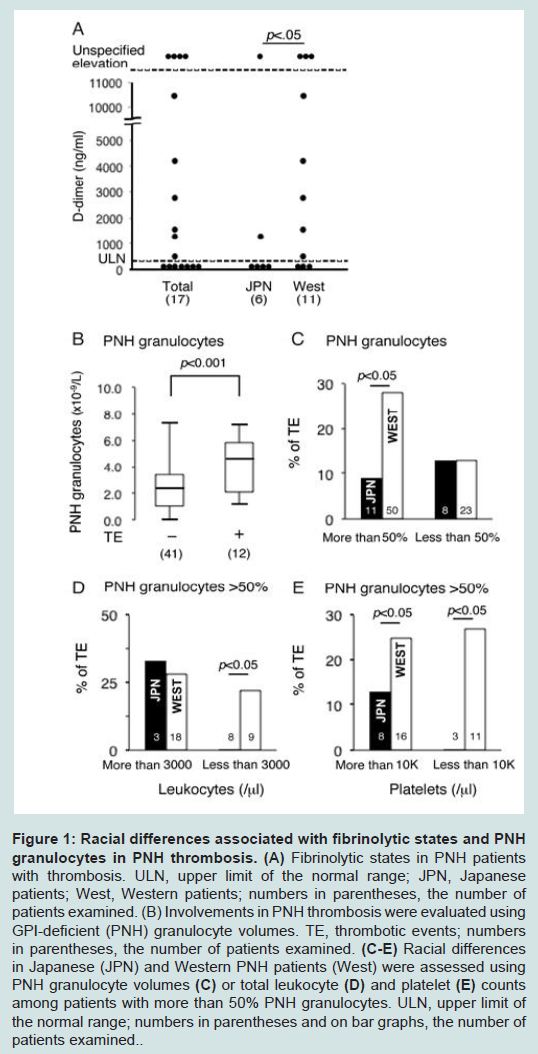
Figure 1: Racial differences associated with fibrinolytic states and PNH granulocytes in PNH thrombosis. (A) Fibrinolytic states in PNH patients with thrombosis. ULN, upper limit of the normal range; JPN, Japanese patients; West, Western patients; numbers in parentheses, the number of patients examined. (B) Involvements in PNH thrombosis were evaluated using GPI-deficient (PNH) granulocyte volumes. TE, thrombotic events; numbers in parentheses, the number of patients examined. (C-E) Racial differences in Japanese (JPN) and Western PNH patients (West) were assessed using PNH granulocyte volumes (C) or total leukocyte (D) and platelet (E) counts among patients with more than 50% PNH granulocytes. ULN, upper limit of the normal range; numbers in parentheses and on bar graphs, the number ofpatients examined.
Acknowledgements
N.H. designed and performed the research, analyzed data, and wrote the manuscript. T.M., S.M., S.N., K.H., T.K., and T.S. analyzed clinical data. H.N. supervised the project, analyzed data, and wrote the manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Ministry of Labour and Welfare of Japan, and the Takeda Science Foundation.References
- Kanakura Y, Ohyashiki K, Shichishima T, Okamoto S, Ando K, et al. (2011) Safety and efficacy of the terminal complement inhibitor eculizumab in Japanese patients with paroxysmal nocturnal hemoglobinuria: the AEGIS clinical trial. Int J Hematol 93: 36-46.
- Helley D, de Latour RP, Porcher R, Rodrigues CA, Galy-Fauroux I, et al. (2010) Evaluation of hemostasis and endothelial function in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Haematologica 95: 574-581.
- Weitz IC, Razavi P, Rochanda L, Zwicker J, Furie B, et al. (2012) Eculizumab therapy results in rapid and sustained decreases in markers of thrombin generation and inflammation in patients with PNH independent of its effects on hemolysis and microparticle formation. Thromb Res 130: 361-368.
- Nishimura J, Kanakura Y, Ware RE, Shichishima T, Nakakuma H, et al. (2004) Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore) 83: 193-207.
- Steg PG, Bhatt DL, Wilson PW, D'Agostino R, Ohman EM, et al. (2007) One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 297: 1197-1206.
- Liebman HA, Feinstein DI (2003) Thrombosis in patients with paroxysmal noctural hemoglobinuria is associated with markedly elevated plasma levels of leukocyte-derived tissue factor. Thromb Res 111: 235-238.
- Tichelli A, Socie G, Marsh J, Barge R, Frickhofen N, et al. (2002) Outcome of pregnancy and disease course among women with aplastic anemia treated with immunosuppression. Ann Intern Med 137: 164-172.
- Boschetti C, Fermo E, Bianchi P, Vercellati C, Barraco F, et al. (2004) Clinical and molecular aspects of 23 patients affected by paroxysmal nocturnal hemoglobinuria. Am J Hematol 77: 36-44.
- Ware RE, Hall SE, Rosse WF (1991) Paroxysmal nocturnal hemoglobinuria with onset in childhood and adolescence. N Engl J Med 325: 991-996.
- Noji H, Shichishima T, Okamoto M, Shichishima-Nakamura A, Matsumoto H, et al. (2007) Microvascular thrombosis in the hepatic vein of a patient with paroxysmal nocturnal hemoglobinuria. Int J Hematol 86: 216-221.