
Journal of Parkinsons disease and Alzheimers disease
Download PDF
Review Article
*Address for Correspondence: Nagaendran Kandiah, National Neuroscience Institute, Department of Neurology, 11 Jalan Tan Tock Seng, Singapore 308433, Singapore, Tel: 6563577171; E-mail: Nagaendran_Kandiah@nni.com.sg
Citation: Foo H, Kandiah N. The Role of Cerebrovascular Disease in Parkinson’s Disease Related Cognitive Impairment. J Parkinsons Dis Alzheimer Dis. 2016;3(2): 7.
Copyright © 2016 Foo H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson's disease & Alzheimer's disease| ISSN: 2376-922X | Volume: 3, Issue:2
Submission: 29 July, 2016 | Accepted: 26 September, 2016 | Published: 06 October, 2016

 The importance of WMH should not be undermined in PD: one systemic review emphasized the use of WMH as an indication of increased risk of stroke, dementia, and death [16] while another study has also showed associations between WMH accumulation and greater cognitive dysfunction and reduced gait performance overtime [17]. In addition, another paper has also shown that WMH could be a significant predictor of the conversion from MCI todementia in PD patients [18]. It has also been shown in one study that WMH distribution was more widespread and extensive in PD-MCI compared to PD-NCI patients [19]. Taken together, the prominent role of WMH begets the need to use WMH as an intermediate marker in the diagnostic work-up of PD [19].
The importance of WMH should not be undermined in PD: one systemic review emphasized the use of WMH as an indication of increased risk of stroke, dementia, and death [16] while another study has also showed associations between WMH accumulation and greater cognitive dysfunction and reduced gait performance overtime [17]. In addition, another paper has also shown that WMH could be a significant predictor of the conversion from MCI todementia in PD patients [18]. It has also been shown in one study that WMH distribution was more widespread and extensive in PD-MCI compared to PD-NCI patients [19]. Taken together, the prominent role of WMH begets the need to use WMH as an intermediate marker in the diagnostic work-up of PD [19].
The Role of Cerebrovascular Disease in Parkinson’s Disease Related Cognitive Impairment
Heidi Foo1 and Nagaendran Kandiah1,2*
- 1National Neuroscience Institute, Department of Neurology, Singapore
- 2Duke-NUS Graduate Medical School, Singapore
*Address for Correspondence: Nagaendran Kandiah, National Neuroscience Institute, Department of Neurology, 11 Jalan Tan Tock Seng, Singapore 308433, Singapore, Tel: 6563577171; E-mail: Nagaendran_Kandiah@nni.com.sg
Citation: Foo H, Kandiah N. The Role of Cerebrovascular Disease in Parkinson’s Disease Related Cognitive Impairment. J Parkinsons Dis Alzheimer Dis. 2016;3(2): 7.
Copyright © 2016 Foo H, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Parkinson's disease & Alzheimer's disease| ISSN: 2376-922X | Volume: 3, Issue:2
Submission: 29 July, 2016 | Accepted: 26 September, 2016 | Published: 06 October, 2016
Abstract
With the high prevalence rate of mild cognitive impairment (MCI) and dementia in Parkinson’s disease (PD), there is an increasing need to establish biomarkers that could identify those at risk of dementia. Small vessel cerebrovascular disease (SVD), including white matter hyperintensities (WMH), lacunes, perivascular spaces (PVS), and microbleeds, has been associated with the motor functions and cognitive impairment in PD. This suggests that SVD could be one of the mechanisms underlying the pathophysiological alterations that occur in the course of PD. Despite its importance, there is a paucity of literature and a lack of consensus in findings on SVD in PD. Without a clear understanding of the interaction between SVD and PD, patient management and care could be compromised. As such, this review summarizes the findings from current literature of SVD in PD, highlighting the limitations that could drive the heterogeneity in results. We also examine the role of other factors, such as microinfarcts and genetics, in their associations with SVD and motor-cognition in PD. Lastly, we discuss the role of optimizing vascular risk factors and the role of antiplatelets in managing patients with concomitant SVD and PD. Consensus on the definition of what constitutes the individual markers of SVD is imperative to determine the method of standardized imaging strategies and for better care management for patients with concomitant SVD and PD.Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder that is typically characterized by its classical presentation of cardinal motor symptoms, including tremors, rigidity, bradykinesia, and postural instability [1]. However, non-motor symptoms, such as cognitive impairment and dementia, are increasingly recognized as a common feature of PD [2,3]. The prevalence of dementia in PD is between 20% and 30% and is associated with increased morbidity and mortality [4]. More significantly, it has been reported that up to 80% of patients with PD progress to mild cognitive impairment (MCI) and dementia (PDD) overtime [5]. Given the multitude of negative consequences associated with PD, it is imperative to establish biomarkers that could identify patients at risk of dementia and for subsequent patient management.Cerebrovascular disease (CVD) has been demonstrated to be associated with cognitive impairment related to PD. Among the different subtypes of CVD, cerebral small vessel disease (SVD) is a term commonly used to describe all pathological processes that affect the perforating cerebral arterioles, capillaries, and venules resulting in atrophy in the cerebral white and deep gray matter [6-8]. Specifically, the ischaemic components of the pathological process of SVD, namely white matter hyperintensities (WMH), chronic lacunar infarcts, perivascular spaces (PVS), and microbleeds [6-9], have been implicated in the manifestations of motor and non-motor symptoms in PD [10,11]. One study has shown that brains of PD patients that lacked evidence of a specific neurodegenerative process demonstrated significantly more severe SVD-associated pathology compared to controls, which suggests that SVD may have a role in the pathophysiology of PD [12]. Another longitudinal study by Foo et al. demonstrated that progression of SVD was associated with significant cortical thinning in the frontoparietal regions with concomitant decline in memory, executive functions, and motor functions in PD patients [13]. This implies that the extent and progression of SVD could be associated with focal cerebral atrophy and domain-specific cognitive dysfunction. Furthermore, it could be possible that concomitant SVD in PD could alter the clinical-cognitive symptom presentation and treatment response of PD [14]. Additionally, Arena et al. showed that deep WMH burden was not only associated with worse performance on UPDRS, such as speech, facial expression, neck rigidity, postural stability, posture, and global spontaneous movements, but also worse response to L-Dopa [15]. This has important clinical relevance in patient management. Presence of WMH may impact response to L-dopa treatment and deep brain stimulation surgery (DBS), since evidence has established the correlations between response to L-Dopa and DBS [15]. Despite the consequence of SVD in PD, there is a paucity of literature investigating the impact of SVD as a whole in the pathophysiology of PD. Even within those who examined SVD in PD, the findings have been heterogeneous. This could be a result of the challenges to image and investigate small vessels in vivo, especially when imaging of small vessel disease are typically measured using subjective visual ratings instead of objective volumetric analysis; and the imprecise and inconsistent methods of classifying the lesions that could lead to under or over diagnosis of SVD [8]. In turn, it is imperative to establish the contributions of SVD to the underlying neuropathophysiology of PD in order to benefit subsequent patient management and treatment.
In this review, we provide the definition of the individual markers of SVD, suggest the importance of each in affecting the pathophysiology of PD, outline the current and emerging concepts of each SVD marker in relation to PD, and highlight the challenges faced in the present literature. We also examine other factors, such as microinfarcts and genetics, and their associations with SVD and clinical-cognitive functions. We then conclude by suggesting potential management strategies (Figure 1).
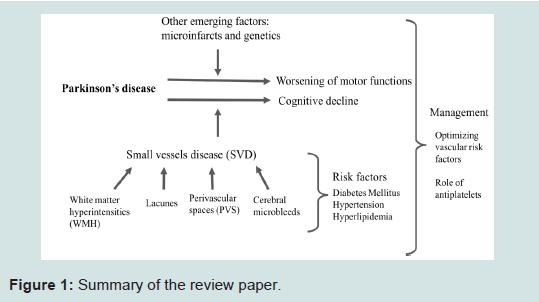
Figure 1: Summary of the review paper.
White Matter Hyperintensities and PD
White matter hyperintensities (WMH) are abnormal areas of increased signal intensity on T2-weighted and fluid attenuated inversion recovery (FLAIR) images (Figure 2) [8]. Often, WMH are distributed in the periventricular and deep white matter of the cerebral hemispheres, in the basal ganglia and pons, and occasionally in the brainstem and cerebellar white matter [8].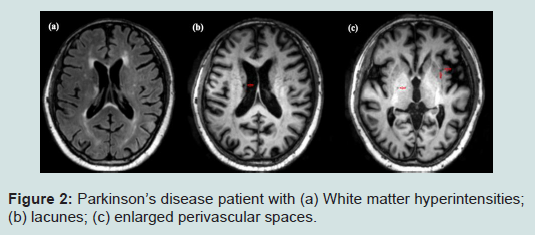
Figure 2: Summary of the review paper.
While WMH has been one of the more prominent feature of SVD that has been studied in the PD literature, there is still heterogeneity in findings - consensus as to whether WMH has a compounded effect on the PD pathology has yet to be fully elucidated. Previous studies have also shown the associations between WMH and motor symptoms [4,17]. Whereas de Laat et al. demonstrated the WMH, specifically in the frontoparietal cortices, were associated with increased risk of parkinsonian signs [4], Silbert et al. showed that periventricular WMH was associated with decreased gait performance overtime [17]. Although there is general consensus that WMH has an effect on the presentation and severity of symptoms in PD, Dalaker et al. did not find similar results [20,21]. In their study, no significant differences were seen in whole brain volume and total WMH volume in PD patients compared to age-matched healthy controls, and associations between cognition and MRI variables did not survive after controlling for confounders [20,21]. While it could be argued that atrophy is not yet widespread in early stages of PD as seen by the results, it could be the corollary of the relatively small sample size that led to the results.
Besides the effects of WMH on motor symptoms in PD, negative associations between WMH and cognition of PD patients have been reported; PD patients with greater cognitive impairment had greater burden of WMH [3,21,22]. In addition, these studies also investigated the burden of WMH on the trajectory of cognitive decline and found that WMH contributed to the progression of dementia in PD patients [3,21,22]. Notably, evidence has demonstrated that PDpatients with high burden of WMH showed poorer performance on frontal-lobe cognitive tasks compared to those with lower WMH; further investigations revealed that deep WMH was closely coupled with cortical thinning in the frontal cortices, which could have led to decline in executive functions [24,21,22]. In all, WMH could potentially have a compounded consequence on the pathogenesis of PD. There are inherent challenges in determining the influence of WMH on the pathophysiology of PD. Firstly, there is a scarcity of longitudinal evidence in the WMH literature within PD, which hinders the interpretation of causality. Additionally, cross-sectional studies may also be less sensitive to subtle changes, which tend to be masked by the large interindividual variability in the brain size andstructure. This warrants the need for more longitudinal studies in order to investigate the progression of WMH and its course on atrophy and clinical-cognitive outcomes. Secondly, few studies investigated regional WMH and concomitant grey matter atrophy in relation to clinical-cognitive outcomes. As Braak and Braak posited that atrophy is not widespread in early stages of PD, there could be localizedatrophy that could be associated with localized WMH regions and in turn, affecting the domain-specific functions [25]. Lastly, the inherent challenge lies in the variability in the quantification of WMH. While some studies used validated semi-quantitative visual ratings, others used volumes; even within each, there are differing scales and methods to quantify WMH. This discrepancy creates mixed findings. Therefore, the current concepts of WMH in PD are limited and future studies should seek to address these limitations. As has been done in vascular dementia, a framework for quantifying and classifying CVD in the context of PD is timely.
Lacunes and PD
Lacunes are small cerebrospinal fluids- (CSF) containing cavities located in the deep grey matter or white matter [8]. To identify lacunes, the combination of the following image acquisitions such as FLAIR, magnetization prepared rapid-acquisition gradient echo (MPRAGE), and T2 are used [26]. Typically, lacunes have ahyperintense rim around the cavity on FLAIR images and hypointense foci on MPRAGE T1 images [6]. However, when present without a hyperintense rim in the basal ganglia and infratentorial regions on T2, they can be distinguished by their shape and size - they are defined as ovoid/spheroid cavities that are 3 mm to 15 mm (Figure 2) [6,8,26]. With evidence showing that striatal lacunar infarcts were three times more common than cortical infarction in pathologically confirmed PD, it could be postulated that lacunes are predominant stroke subtypes in PD [14,27]. Additionally, lacunes on MRI parallel asteeper rate of decline in psychomotor speed and executive functions, which suggests that lacunes play an important role in influencing the clinical-cognitive symptoms in PD. Despite its importance, the clinical significance of lacunes has received much lesser attention as compared to WMH.Evidence has shown that Parkinsonian signs were found more frequently in lacunar than in territorial stroke patients: bradykinesia in 45% and 7%, rigidity in 13% and 7%, tremor in 6% and 7%, and gait disorder in 16% and 7%, respectively [28]. In agreement with this finding, it has also been shown that lacunar infarcts, particularly inthe thalamus, were independently associated with an increased risk of mild parkinsonian signs, mainly due to the presence of bradykinesia [4].
While the clinical relevance of lacunes to motor symptoms is widely accepted, the degree to which cerebral lacunes affect cognition has been less clear [29]. The relative contribution of lacunes to cognitive functions has been contradictory. With uncertainly in previous cross-sectional studies, some showed a significant correlationbetween lacunes and cognitive symptoms [30,31] while others did not [32,33]. The few longitudinal studies have result in mixed findings [34-37]. Investigation of the associations between the presence of lacunes and cognitive dysfunction in the elderly population is paramount as it sheds light into the possible relationship they have in the PD population. Findings from the elderly population showed that cognitive performance was significantly poorer in elderly subjects with lacunes across cognitive domains such as memory, processing speed, motor functions, and executive functions compared to those without lacunes [38,39]. It has been posited that the mechanisms underlying the subcortical lacunes and executive dysfunction may be related to cortical hypometabolism: evidence from FDG-PET studies showed that lacunes are associated with frontal hypometabolism, particularly within the prefrontal cortex, which could explain the poorer executive functions [39,40]. Similar results were also seen in ongitudinal studies [34,35]. Taken together, lacunes could contribute to impairment in different cognitive domains and identifying individuals with subclinical vascular brain changes could result in improved understanding of cognitive impairment in old age and potentially in PD [38].
The challenges in studying lacunes in PD patients include the definition of lacunes and the number of lacunes present to make considerable motor and cognitive impact. For the former, the terms lacunar infarcts and lacunes have been used interchangeably eventhough they are different; while lacunes refer to the abovementioned, lacunar infarcts should refer to a clinical stroke syndrome of lacunar type where the underlying lesion is an infarct on brain imaging [8]. The differentiation of these terms should be clear in order to specifically determine the vascular pathology that interacts with the neurodegenerative process of PD that results in motor and cognitive dysfunction. The latter is an issue as while dementia associated with multiple lacunes has been widely recognized, cognitive dysfunction resulting from one or few lacunes is often considered negligible [41]. This is an important issue that should be acknowledged based on the vidence that even a single lacunecan produce cognitive deficits [42].
Perivascular Spaces and PD
Perivascular spaces (PVS), are fluid-filled spaces that are generally smaller than 3 mm in diameter, which surround the deep perforating arterioles as the arterioles pass through the deep grey and whitematter (Figure 2) [8,43]. On MRI, these spaces are visible on either T2-weighted or T1 images as they have similar signal intensity similar to CSF [43]. As they follow the course of the vessel, they appear round in the basal ganglia and linear in the subcortical white matter of lateral parts of the lobes on axial images [43].PVS have been postulated to have an important role in the brain’s inflammatory and immunological response [41]. Rameirez et al. hypothesized the clearance pathways in explaining the essential role of PVS. Clearance of metabolic waste and fluid from the brain parenchyma may occur via various pathways, including degradationand clearance through the blood brain barrier, PVS, and CSF [44,45]. Collectively, these pathways maintain homeostasis within the brain. However, the contribution and importance of each pathway remains to be understood [41]. With disease, dysfunction of one or more of these pathways may result in further pathological compromise and further disrupting the removal of toxins and metabolic waste from the brain. As such, possibly, one of the pathological hallmark of compromised fluid and toxic clearance from the brain may be the enlargement of the PVS (ePVS), defined as dilated PVS with diameter equal to or more than 15 mm [47], which could represent a potential biomarker of clearance failure within the central nervous system [44]. Hence, ePVS could serve as a potential biomarker to neurodegenerative diseases, such as PD.
Enlarged PVS have been implicated with motor dysfunction as well as cognitive decline in PD [43,46]. While the exact pathological process leading to dilated PVS is still unknown, they have been associated with arteriolosclerosis, hypertension or other vascular risk factors, dementia, and reduced cognitive functions in elderly [46].While it is still unclear whether ePVS should be recognized as normal or pathological ageing [47], reports from multiple case studies suggested that ePVS could contribute to observed atypical clinical features in patients who otherwise had clinical/imaging findings consistent with PD [48]. In one case study, it was reported that a patient with facial dystonia, chin tremor, and a postural and action-induced dystonic tremor with concomitant ePVS in the substantia nigra had partial response to levodopa dose, suggesting ePVS could also contribute to the postsynaptic changes as seen in this case [49]. In accordance with previous MRI studies, diffusion tensor imaging (DTI) studies, which examines water motion in vivo, showed correlations between changes in the corticospinal tracts and sensorimotor deficits and increased risk of dementia [50,51]. Additionally, findings from a previous study also demonstrated that increased basal ganglia/centrum semiovaleePVS is associated with worse non-verbal reasoning and typically, visuospatial cognitive ability [52].
Despite the promising results of ePVS, it is important to note that the etiology underlying the pathology of PD is probably multifactorial. As such, in order to demonstrate the causal relationship betweenePVS and the clinical-cognitive symptoms in PD, further longitudinal studies possibly with pathological correlations would be encouraged.
Cerebral Microbleeds and PD
Microbleeds are well defined small hypointense punctate on T2* gradient-recalled echo or susceptibility-weighted images that generally less than 10 mm in diameter [8,43]. Normally, they are located in the cortico-subcortical junction, and deep grey or white matter in the cerebral hemispheres, brainstem, and cerebellum [43].With literature demonstrating that cerebral microbleeds are related to underlying cerebral small vessel diseases, increased risk of symptomatic intracerebral hemorrhage, incidence of cognitive impairment and dementia, it could be postulated that microbleedscontribute to the clinical manifestations and cognitive performance of PD patients [53]. In addition, it has been reported that cerebral microbleeds were found in 17.4% and 17.7% of PD patients in United Kingdom and Korea respectively [54,55]. Therefore, there is a growing need to regard microbleeds as a significant precursor to the motor and cognitive decline seen in PD.
Despite the importance of cerebral microbleeds in PD, there is a lack of literature and findings from these studies are also inconclusive. Cross-sectional studies have shown that microbleeds are not associated with motor symptoms in PD: frequencies of motorsubtypes were similar in PD patients with microbleeds and those without; and the average Hoehn and Yahr did not differ based on the presence of microbleeds [54,55]. This suggests that the presence of microbleeds could have no associations with disease severity in PD. However, more studies with larger sample size and recruitment of early stage PD should be conducted in order to confirm the results.
Findings of microbleeds affecting cognition in PD were also mixed. On one hand, some cross-sectional studies found that microbleeds were associated with cognitive decline, typically in the executive function domain [56-60] others did not [54,55]. In addition, microbleeds were seen to occur more frequently in patients with PDD than in those with non-demented PD [61]. Furthermore, evidence has also shown that lobar microbleeds were also associatedwith regions of the brain associated with domain-specific cognitive decline [56]. Hence, microbleeds may contribute to an exacerbation of further cognitive decline in specific domains in PD patients.
More importantly, the presence of the number of microbleeds showed important relationship to cognition: some authors advocate the presence of 2 or more microbleeds in order to find an association with cognition [57] whereas others used 4 [62] or more [59] microbleeds as the benchmark. This inconsistency could generate conflicting results. Future studies should seek to clarify this issue. A longitudinal study is also necessary to clarify if the presence of microbleeds constitute an additive burden of ongoing cognitive dysfunction [61].
Other Factors Associated with SVD
Other factors, such as cerebral microinfarcts and genetics, which relates to SVD and affects motor-cognition symptoms in PD have not been fully explored. These are potential factors that if elucidated could have consequential impact on the management care of PD patients.Cerebral microinfarcts are lesions that often result from small vessel pathologies such as arteriolosclerosis and cerebral amyloid angiopathy (CAA). Typically, microinfarcts go undetected in clinical radiological studies relying on conventional structural imaging as they are very small lesions with approximately 0.2 mm in diameter [63]. Furthermore, thus far, the ex-vivo studies’ examining microinfarcts have been cross-section and therefore, limits their ability to explore the incidence and consequence of microinfarcts [64-66]. Despite their limitations, these ex-vivo studies have postulated that the presence of numerous microinfarcts may be capable of causing clinical ymptoms, specifically cognitive dysfunction, while accounting for other pathologies including macroinfarcts and lacunes [64-66]. However, it is important to note that the sample from these studies usually represent patients in the intermediate AD stage, which is not representative of the general population. In community studies, findings showed that cortical microinfarcts were significantly associated with episodic memory, semantic memory, and visual perception speed and not with subcortical mircoinfarcts [67]. In addition, the few studies investigating microinfarcts in Parkinsonism showed that the presence of multiple infarcts has been linked to gait impairment [68-70]. Taken together, there are promising results in relating microinfarcts to cognitive dysfunction as well as motor symptoms, however, there needs to be studies in larger longitudinal PD cohorts in order to confirm these results.
While there is some literature associating genetics with SVD, the exact pathogenesis is still uncertain. Results from one candidate gene study, which is an approach involving exploring genetic influence on complex trait by identifying candidate genes that might have a role in the etiology of the disease, showed that there were no associationsbetween genes such as apolipoprotein E (ApoE) (e4+/-) [71]. However, this is in contrast with the established literature that the ApoE genotype relates to an increased risk of developing sporadic CAA, which could imply the underlying degenerative vascular changes similar to small vessels disease [72]. The variability in results suggests the need to study ApoE in a longitudinal large cohort. Moreover, novel genotyping methods have also showed promising results. One of the methods is the use of the genome-wide association study (GWAS), which measures and analyzes deoxyribonucleic acid (DNA) sequence variations from across the human genome in attempts to identify genetic risk factors for diseases and the biological underpinnings of disease susceptibility for developing prevention and treatment strategies [73]. One GWAS study has demonstrated that single nucleotide polymorphisms (SNPs) rs3744028 and rs1055129 contributed to cerebral WMH, however only about 4% to 8% of the overall mean WMH burden in the sample [74]. Although several SNPs have been identified, detailed information on the specific genes and functional variants should be investigated further to determine the exact role of the gene function in the underlying pathogenic mechanisms of SVD [75]. Other genetics of single gene disorders that cause cerebral SVD include CADASIL caused by mutations in the NOTCH3 gene on chromosome 19q12 and has been implicated in progressive cognitive decline [76,77]. Hence, these different genetictechniques have shown promising results in relating genetics to SVD. Future studies should look into the underlying pathogenesis of genetics of SVD in PD cohort. This could have important implications in clinical presentation and cognitive performance of PD patients and so influence the management of these patients.
Management
Therapeutic options in patients with concomitant SVD in PD are scarce. In this review, we evaluate a few management strategies, such as the role of optimizing vascular risk factors and the role of antiplatelets in cognition in PD, that have been reported to be useful in prevention of dementia and management of these patients.Although the exact mechanisms of the individual markers of SVD differ, they have been associated with vascular risk factors, such as diabetes mellitus, hypertension, and hyperlipidemia, which ave consequential implications on the cognitive status of patients with PD. As The Rotterdam study has reported that up to one-thirdof dementia cases could be prevented through optimal prevention and treatment of vascular risk factors, similar trend could also be projected onto patients with cognitive dysfunction in PD [78].Community-based studies have consistently demonstrated the associations between WMH and hypertension [79]. It has also been reported that effective hypertension treatment was associated with lower risk of WMH; reduction in dementia cases were found in subjects treated for isolated systolic hypertension [80,81]. In addition, another study has also shown correlations that smaller lacunes were associated with diabetes mellitus whereas larger oneswere associated with low-density lipoprotein (LDL) cholesterol suggesting that diabetes could be associated with fibrinoid necrosis and LDL cholesterol with microatheroma that have been implicated in the risk of lacunes [82]. One study has shown that by lowering the prevalence of diabetes by 25%, more than 200,000 dementia cases could potentially be prevented worldwide [83]. In the same respect, by managing diabetes in PD patients, cognitive impairment and dementia cases within PD could be reduced. Similarly, microbleeds have also showed correlations with systolic blood pressure and severe hypertension [84]. Taken together, managing potentially modifiable vascular risk factors could potentially lower the incidence of cognitive dysfunction in PD.Antiplatelet drugs, such as aspirin and cilostazol, may also be beneficial to patients with SVD. Studies have demonstrated that vascular dementia and dementia patients on antiplatelet treatmentwere at lower risk of death [85]. Additionally, in a randomized clinical trial of low dose aspirin, it showed that aspirin played an important role in reducing the risk of myocardial infarction and stroke, and slowing the onset and progression of dementia [86]. Moreover, cilostazol has also been suggested to decrease betaamyloid accumulation and protect beta-amyloid induced cognitive deficits [87]. Specifically, one study has shown that cilostazol seemedto be safer than aspirin in terms of the risk of hemorrhagic stroke in hypertensive patients with SVD [88]. However, the use of antiplatelet begets the problem of degree of benefit/risk ratio in patients with vascular dementia according to the risk of hemorrhage, which isincreased by the presence of silent microhemorrhages on MRI frequently detected in the presence of ischemic white matter lesions and by gait disturbances and falls [89]. Despite so, due to the relatively low efficacy of available drugs to improve cognition in patients with SVD, which may lead to the development of dementia, antiplatelet drugs could be one of the key treatments to managing patients with SVD in PD and in minimizing the risk of dementia in these patients.
Conclusion
In conclusion, there is increasing evidence linking measures of SVD, such as WMH, lacunes, PVS, and microbleeds to motor andcognitive symptoms in PD. However, the lack of uniform criteria and standardized measurement techniques continue to hamper progress in research related to CVD and PD. As such, there is an urgent need to develop consensus criteria in order to ensure standardized imaging techniques to determine SVD and also for better patient management strategies to move this field forward.References
- Gelb DJ, Oliver E, Gilman S (1999) Diagnostic criteria for Parkinson disease. Arch Neurol 56: 33-39.
- Christopher L, Strafella AP (2013) Neuroimaging of brain changes associated with cognitive impairment in Parkinson’s disease. J Neuropsychol 7: 225-240.
- Kandiah N, Mak E, Ng A, Huang S, Au WL, et al. (2013) Cerebral white matter hyperintensity in Parkinson’s disease: a major risk factor for mild cognitive impairment. Parkinsonism Relat Disord 19: 680-683.
- de Laat KF, van Norden AG, Gons RA, van Uden IW, Zwiers MP, et al. (2012) Cerebral white matter lesions and lacunar infarcts contribute to the presence of mild parkinsonian signs. Stroke 43: 2574-2579.
- Mak E, Su L, Williams GB, O’Brien JT (2015) Neuroimaging correlates of cognitive impairment and dementia in Parkinson’s disease. Parkinsonism Relat Disord 21: 862-870.
- Pantoni L (2010) Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 9: 689-701.7.
- Staals J, Booth T, Morris Z, Bastin ME, Gow AJ, et al. (2015) Total MRI load of cerebral small vessel disease and cognitive ability in older people. Neurobiol Aging 36: 2806-2811.
- Wardlaw JM, Smith C, Dichgans M (2013) Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 12: 483-497.
- Tuladhar AM, van Dijk E, Zwiers MP, van Norden AG, de Laat KF, et al. (2016) Structural network connectivity and cognition in cerebral small vessel disease. Hum Brain Mapp 37: 300-310.
- Hatate J, Miwa K, Matsumoto M, Sasaki T, Yagita Y, et al. (2016) Association between cerebral small vessel diseases and mild parkinsonian signs in the elderly with vascular risk factors. Parkinsonism Relat Disord 26: 29-34.11.
- van der Holst HM, van Uden IW, Tuladhar AM, de Laat KF, van Norden AG, et al. (2015) Cerebral small vessel disease and incident parkinsonism: the RUN DMC study. Neurology 85: 1569-1577.
- Zijlmans J, Evans A, Fontes F, Katzenschlager R, Gacinovic S, et al. (2007) [123I] FP-CIT spect study in vascular parkinsonism and Parkinson’s disease. Mov Disord 22: 1278-1285.
- Foo H, Mak E, Yong TT, Wen MC, Chander RJ, et al. (2016) Progression of small vessel disease correlates with cortical thinning in Parkinson’s disease. Parkinsonism Relat Disord [Epub ahead of print].14.
- Schwartz RS, Halliday GM, Cordato DJ, Kril JJ (2012) Small-vessel disease in patients with Parkinson’s disease: a clinicopathological study. Mov Disord 27: 1506-1512.
- Arena JE, Cerquetti D, Rossi M, Chaves H, Rollan C, et al. (2016) Influence of white matter MRI-hyper-intensities on acute l-dopa response in patients with Parkinson’s disease. Parkinsonism Relat Disord 24: 126-128.16.
- Debette S, Markus HS (2010) The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: systemic review and meta-analysis. BMJ 341: c3666.
- Silbert LC, Nelson C, Howieson DB, Moore MM, Kaye JA (2008) Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline. Neurology 71: 108-113.18.
- Kandiah N, Zanial NH, Narasimhalu K, Chander RJ, Ng A, et al. (2014) Hippocampal volume and white matter disease in the prediction of dementia in Parkinson’s disease. Parkinsonism Relat Disord 20: 1203-1208.
- Mak E, Dwyer MG, Ramasamy DP, Au WL, Tan LC, et al. (2015) White matter hyperintensities and mild cognitive impairment in Parkinson’s disease. J Neuroimaging 25: 754-760.
- Dalaker TO, Larsen JP, Bergsland N, Beyer MK, Alves G, et al. (2009) Brain atrophy and white matter hyperintensities in early parkinson’s disease(a). Mov Disord 24: 2233-2241.21.
- Dalaker TO, Larsen JP, Dwyer MG, Aarsland D, Beyer MK, et al. (2009) White matter hyperintensities do not impact cognitive function in patients with newly diagnosed Parkinson’s disease. Neuroimage 47: 2083-2089.
- Lee SJ, Kim JS, Yoo JY, Song IU, Kim BS, et al. (2010) Influence of white matter hyperintensities on the cognition of patients with parkinson disease. Alzheimer Dis Assoc Disord 24: 227-233.
- Sunwoo MK, Jeon S, Ham JH, Hong JY, Lee JE, et al. (2014) The burden of white matter hyperintensities is a predictor of progressive mild cognitive impairment in patients with Parkinson’s disease. Eur J Neurol 21: 922-e50.24.
- Ham JH, Yun HJ, Sunwoo MK, Hong JY, Lee JM, et al. (2015) Topography of cortical thinning associated with white matter hyperintensities in Parkinson’s disease. Parkinsonism Relat Disord 21: 372-377.25.
- Braak H, Braak E (2000) Pathoanatomy of Parkinson’s disease. J Neurol 247 Suppl 2: II3-II10.
- Gouw AA, van der Flier WM, Fazekas F, van Straaten EC, Pantoni L, et al. (2008) Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period; the leukoraiosis and disability study. Stroke 39: 1414-1420.27.
- Hughes AJ, Daniel SE, Blankson S, Lees AJ (1993) A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 50: 140-148.
- van Zagten M, Lodder J, Kessels F (1998) Gait disorder and parkinsonian signs in patients with stroke related to small deep infarcts and white matter lesions. Mov Disord 13: 89-95.29.
- Filley CM, Brodtmann A (2011) Lacunes and cognitive decline: little things matter. Neurology 76: 1856-1857.
- Gold G, Kovari E, Herrmann FR, Canuto A, Hof PR, et al. (2005) Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 36: 1184 -1188.
- Reitz C, Trenkwalder C, Kretzschmar K, Roeslr A, Eckardstein AV, et al. (2006) Relation of cerebral small-vessel disease and brain atrophy to mild parkinsonism in elderly. MovDisord 21: 1914-1919.32.
- Fein G, Di Sclafani V, Tanabe J, Cardenas V, Weiner MW, et al. (2000) Hippocampal and cortical atrophy predict dementia in subcortical ischemic vascular disease. Neurology 55: 1626 -1635.33.
- Mungas D, Jagust WJ, Reed BR, Kramer JH, Weiner MW, et al. (2001) MRI predictors of cognition in subcortical ischemic vascular disease and Alzheimer’s disease. Neurology 57: 2229 -2235.34.
- Jokinen H, Gouw AA, Madureira S, Ylikoski R, van Straaten EC, et al. (2011) Incident lacunes influences cognitive decline: the LADIS study. Neurology 76: 1872-1878.
- Knopman DS, Penman AD, Catellier DJ, Coker LH, Shibata DK, et al. (2011) Vascular risk factors and longitudinal changes on brain MRI: the ARIC study. Neurology 76: 1879-1885.36.
- Mungas D, Harvey D, Reed BR, Jagust WJ, DeCarli C, et al. (2005) Longitudinal volumetric MRI change and rate of cognitive decline. Neurology 65: 565-571.
- van Dijk EJ, Prins ND, Vrooman HA, Hofman A, Koudstaal PJ, et al. (2008) Progression of cerebral small vessel disease in relation to risk factors and cognitive consequences: Rotterdam scan study. Stroke 39: 2712-2719.38.
- Baune BT, Roesler A, Knecht S, Berger K (2009) Single and combined effects of cerebral white matter lesions and lacunar infarctions on cognitive function in an elderly population. J Gerontol A Biol Sci Med Sci 64: 118-124.39.
- Carey CL, Kramer JH, Josephson SA, Mungas D, Reed BR, et al. (2008) Subcortical lacunes are associated with executive dysfunction in cognitively normal elderly. Stroke 39: 397-402.
- Reed BR, Eberling JL, Mungas D, Weiner M, Jagust WJ (2001) Frontal lobe hypometabolism predicts cognitive decline in patients with lacunar infarcts. Arch Neurol 58: 493-497.41.
- Fisher CM (1982) Lacunar strokes and infarcts: a review. Neurology 32: 871-876.
- van Zandvoort MJ, van der Grond J, Kapelle LJ, de Haan EH (2005) Cognitive deficits and changes in neurometabolites after a lacunar infarct. J Neurol 252: 183-190.43.
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, et al. (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: 822-838.
- Ramirez J, Berezuk C, McNeely AA, Gao F, McLaurin J, et al. (2016) Imaging the perivascular space as a potential biomarker of neurovascular and neurodegenerative diseases. Cell Mol Neurobiol 36: 289-299.45.
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, et al. (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11: 457-470.46.
- Cerase A, Vallone IM, Muccio CF, Petrini C, Signori G, et al. (2010) Regression of dilated perivascular spaces of the brain. Surg Radiol Anat 32: 555-561.
- Potter GM, Doubal FN, Jackson CA, Chappell FM, Sudlow CL, et al. (2015) Enlarged perivascular spaces and cerebral small vessel disease. Int J Stroke 10: 376-381.
- Mestre TA, Armstrong MJ, Walsh R, Dakheel AA, Maco E, et al. (2014) Can isolated enlarged virchow-robin spaces influence the clinical manifestations of Parkinon’s disease? Mov Disord Clin Pract 1: 67-69.
- Mehta SH, Nicholas FT 3rd, Espay AJ, Duker AP, Morgan JC, et al. (2013) Dilated Vichrow-Robin spaces and parkinosonism. Mov Disord 26: 589-599.50.
- Laundre BJ, Jellison BJ, Badie B, Alexander AL, Field AS (2005) Diffusion tensor imaging of the corticospinal tract before and after mass resection as correlated with clinical motor findings: preliminary data. AJNR Am J Neuroradiol 26: 791-796.
- Zhu LL, Lindenberg R, Alexander MP, Schlaug G (2010) Lesion load of the corticospinal tract predicts motor impairment in chronic stroke. Stroke 41: 910-915.
- Maclullich AM, Wardlaw JM, Ferguson KJ, Starr JM, Seckl JR, et al. (2004) Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry 75: 1519-1523.53.
- Cai Z, Wang C, He W, Tu H, Tang Z, et al. (2015) Cerebral small vessel disease and Alzheimer’s disease. Clin Interv Aging 10: 1695-1704
- Yamashiro K, Tanaka R, Hoshino Y, Hatano T, Nishioka K, et al. (2015) The prevalence and risk factors of cerebral microbleeds in patients with Parkinson’s disease. Parkinsonism Relat Disord 21: 1076-1081.
- Kim JH, Park J, Kim YH, Ma HI, Kim YJ (2015) Characterization of cerebral microbleeds in idiopathic Parkinson's disease. Eur J Neurol 22: 377-383.56.
- Gorelick PB, Farooq MU (2016) Cerebral microbleeds, cognition, and therapeutic implications. JAMA Neurol 73: 908-910.
- Hilal S, Saini M, Tan CS, Catindig JA, Koay WI, et al. (2014) Cerebral microbleeds and cognition: the epidemiology of dementia in Singapore study. Alzheimer Dis Assoc Disord 28: 106-112.
- Patel B, Lawrence AJ, Chung AW, Rich P, Mackinn on AD, et al. (2013) Cerebral microbleeds and cognition in patients with symptomatic small vessel disease. Stroke 44: 356-361.
- Poels MM, Ikram MA, van der Lugt A, Hofman A, Niessen WJ, et al. (2012) Cerebral microbleeds are associated with worse cognitive function: the Rotterdam Scan Study. Neurology 78: 326-333.
- Werring DJ, Frazer DW, Coward LJ, Losseff NA, Watt H, et al. (2004) Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain 127(Pt 10): 2265-2275.
- Ham JH, Yi H, Sunwoo MK, Hong JY, Sohn YH, et al. (2014) Cerebral microbleeds in patients with Parkinson’s disease. J Neurol 261: 1628-1635.
- Akoudad S, Wolters FJ, Viswanathan A, de Bruijn RF, van der Lugt A, et al. (2016) Association of cerebral microbleeds with cognitive decline and dementia. JAMA Neurol 73: 934-943.
- Smith EE, Schneider JA, Wardlaw JM, Greenberg SM (2012) Cerebral microinfarcts: the invisible lesions. Lancet Neurol 11: 272-282.
- Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, et al. (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59: 931-945.
- Haglund M, Passant U, Sjobeck M, Ghebremedhin E, Englund E (2006) Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 21: 681-687.66.
- Del Ser T, Hachinski V, Merskey H, Munoz DG (2005) Alzheimer’s disease with and without cerebral infarcts. J Neurol Sci 231: 3-11.
- Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA (2011) Microinfarct pathology, dementia, and cognitive systems. Stroke 42: 722-727.68.
- Kaplan JG, Katzman R, Horoupian DS, Fuld PA, Mayeux R, et al. (1985) Progressive dementia, visual deficits, amyotrophy, and microinfarcts. Neurology 35: 789-796.69.
- Baloh RW, Vinters HV (1995) White matter lesions and disequilibrium in older people. II. Clinicopathologic correlation. Arch Neurol 52: 975-981.
- Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA (2011) Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke 42: 3183-3189.
- Paternoster L, Chen W, Sudlow CL (2009) Genetic determinants of white matter hyperintensities on brain scans: a systematic assessment of 19 candidate gene polymorphisms in 46 studies in 19,000 subjects. Stroke 40: 2020-2026
- Fornage M, Debette S, Bis JC, Schmidt H, Ikram MA, et al. (2011) Genome-wide association studies of cerebral white matter lesion burden: the CHARGE consortium. Ann Neurol 69: 928-939.
- Bush WS, Moore JH (2012) Chapter 11: Genome-wide association studies. PLoS Comput Biol 8: e1002822.
- Choi JC (2015) Genetics of cerebral small vessel disease. J Stroke 17: 7-16.
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, et al. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383: 707-710.
- Choi JC (2010) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a genetic cause of cerebral small vessel disease. J Clin Neurol 6: 1-9.
- Knudsen KA, Rosand J, Karluk D, Greenberg SM (2001) Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 56: 537-539.
- Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM (2009) Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry 80: 13-17
- Jeerakathil T, Wolf PA, Beiser A, Massaro J, Seshadri S, et al. (2004) Stroke risk profile predicts white matter hyperintensity volume: the Framingham study. Stroke 35: 1857-1861.80.
- de Leeuw FE, de Groot JC, Oudkerk M, Witteman JC, Hofman A, et al. (2002) Hypertension and cerebral white matter lesions in a prospective cohort study. Brain 125: 765-772.
- Forette F, Seux ML, Staessen JA, Thijs L, Birkenhäger WH, et al. (1998) Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet 352: 1347-1351.
- Bezerra DC, Sharrett AR, Matsushita K, Gottesman RF, Shibata D, et al. (2012) Risk factors for lacune subtypes in the atherosclerosis risk in communities (ARIC) study. Neurology 78: 102-108.83.
- Barnes DE, Yaffe K (2011) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10: 819-828.
- Poels MM, Ikram MA, van der Lugt A, Hofman A, Krestin GP, et al. (2011) Incidence of cerebral microbleeds in the general population: the Rotterdam scan study. Stroke 42: 656-661.
- Freels S, Nyenhuis DL, Gorelick PB (2002) Predictors of survival in African American patients with AD, VaD, or stroke without dementia. Neurology 59: 1146-1153.
- Nelson M, Reid C, Beilin L, Donnan G, Johnston C, et al. (2003) Rationale for a trial of low-dose aspirin for the primary prevention of major adverse cardiovascular events and vascular dementia in the elderly. Drugs Aging 20: 897-903.87.
- Park SH, Kim JH, Bae SS, Hong KW, Lee DS, et al. (2011) Protective effect of the phosphodiesterase III inhibitor cilostazol on amyloid β-induced cognitive deficits associated with decreased amyloid β accumulation. Biochem Biophys Res Commun 408: 602-608.88.
- Uchiyama S, Shinohara Y, Katayama Y, Yamaguchi T, Handa S, et al. (2014) Benefit of cilostazol in patients with high risk of bleeding: subanalysis of cilostazol stroke prevention study 2. Cerebrovasc Dis 37: 296-303.89.
- Chabriat H, Bousser MG (2006) Vascular dementia: potential of antiplatelet agents in prevention. Eur Neurol 55: 61-69.