
Journal of Syndromes
Download PDF
Case Report
*Address for Correspondence: Marcia Ramos-e-Silva, MD, PhD, Sector of Dermatology and Post-Graduation Course in Dermatology, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Rua Dona Mariana 149/C-32, 22280-020 Rio de Janeiro, Brazil, Tel: 55-21-22864632; E-mail: ramos.e.silva@dermato.med.br
Citation: de Andrade FC, Azulay DR, de Matos Pinto de Carvalho FN, Cuzzi T, Ramos-e-Silva M. Cowden Syndrome: Case Report with Late Diagnosis. J Syndromes. 2015;2(2): 3.
Copyright © 2015 Ramos-e-Silva et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 2, Issue: 2
Submission: 17 August 2015 | Accepted: 31 October 2015 | Published: 05 November 2015



The examination of the oral cavity showed multiple normochromic papules on the labial and buccal mucosa (Figure 4). Additionally, the patient presented evident psychomotor development impairment. The histopathological exam of the skin lesion on the nasal dorsum was compatible with trichilemmoma (Figures 5 and 6). The patient was therefore with the clinical established criteria diagnosed with Cowden syndrome.



There is a wide spectrum of clinical manifestations of the syndrome. Mucocutaneous lesions are present in nearly 100% of patients and manifest as lichenoid papules, acral keratoses, translucent keratosis on the palms, and papillomatous or mixed verrucous lesions. Trichilemmomas, normochromic papules located on the head and neck, are benign hamartomatous lesions derived from the outer sheath of the hair follicle (trichilemma) and are characteristic of the syndrome. Involvement of the mucosa is present in 80% of patients with the most common sites being the buccal mucosa and gingiva, in which the lesions may coalesce with an aspect of pavement stones [6].
Cowden Syndrome: Case Report with Late Diagnosis
Felipe Cupertino de Andrade1, David Rubem Azulay2, Felipe Nazareth de Matos Pinto de Carvalho1, Tullia Cuzzi3 and Marcia Ramos-e-Silva4*
- 1Sector of Dermatology and Post-Graduation Course, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Brazil
- 2Sector of Dermatology, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 3Sector of Pathology, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 4Sector of Dermatology and Post-Graduation Course in Dermatology, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
*Address for Correspondence: Marcia Ramos-e-Silva, MD, PhD, Sector of Dermatology and Post-Graduation Course in Dermatology, University Hospital and School of Medicine, Federal University of Rio de Janeiro, Rua Dona Mariana 149/C-32, 22280-020 Rio de Janeiro, Brazil, Tel: 55-21-22864632; E-mail: ramos.e.silva@dermato.med.br
Citation: de Andrade FC, Azulay DR, de Matos Pinto de Carvalho FN, Cuzzi T, Ramos-e-Silva M. Cowden Syndrome: Case Report with Late Diagnosis. J Syndromes. 2015;2(2): 3.
Copyright © 2015 Ramos-e-Silva et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Syndromes | ISSN: 2380-6036 | Volume: 2, Issue: 2
Submission: 17 August 2015 | Accepted: 31 October 2015 | Published: 05 November 2015
Abstract
The authors present a case of Cowden syndrome, which was only diagnosed at the age of 45. It is very important to make an early diagnosis, periodical clinical and lab exams in these patients in order to prevent the patients from malignant diseases in untreatable situations.Keywords
Cowden syndrome; Trichilemmoma; Oral; MouthIntroduction
Cowden syndrome, also known as multiple hamartomas syndrome, is an autosomal dominant genodermatosis, which affects multiple organs. It is characterized by the presence of hamartomas of ectodermal, mesodermal and endodermal origin, with an increased risk of malignant transformation [1,2]. The authors report a case of late diagnosis of the classic clinical manifestations of this syndrome in a man with exuberant skin lesions.Case Report
Our patient is a 45 year-old white male, born in Rio de Janeiro. He was admitted to our Hospital by the Sector of Endocrinology due to thyroid enlargement and presence of palpable nodules, which did not respond to medication. Thyroidectomy was performed and the histopathological examination revealed papilliferous thyroid carcinoma, multiple bilateral follicular adenomas and multinodular goiter. During hospitalization, skull MRI and video ileocolonoscopy were requested, both being normal. Due to the presence of skin lesions, the patient was consulted at the Sector of Dermatology. Dermatologists observed macrocephaly and multiple normochromic papules, some flat, others keratotic and filiform, tending to form clusters, distributed around ears, nose, eyes and mouth (Figures 1-3).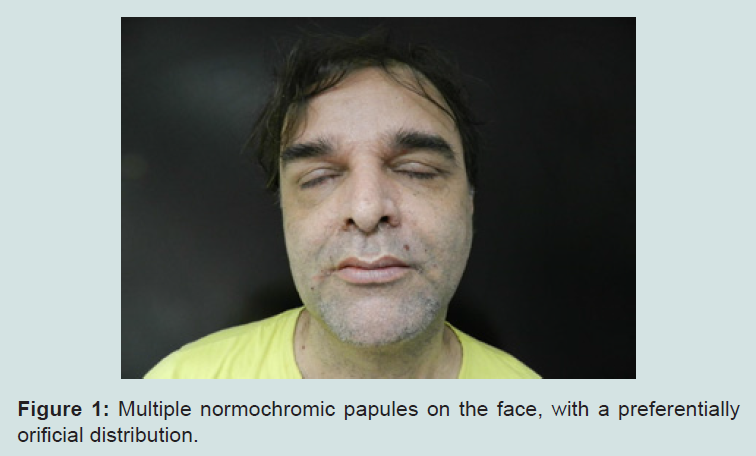
Figure 1: Multiple normochromic papules on the face, with a preferentially orificial distribution.
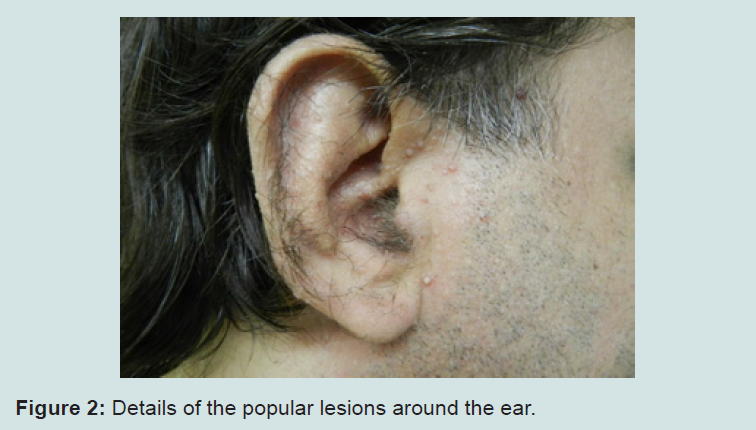
Figure 2: Details of the popular lesions around the ear.
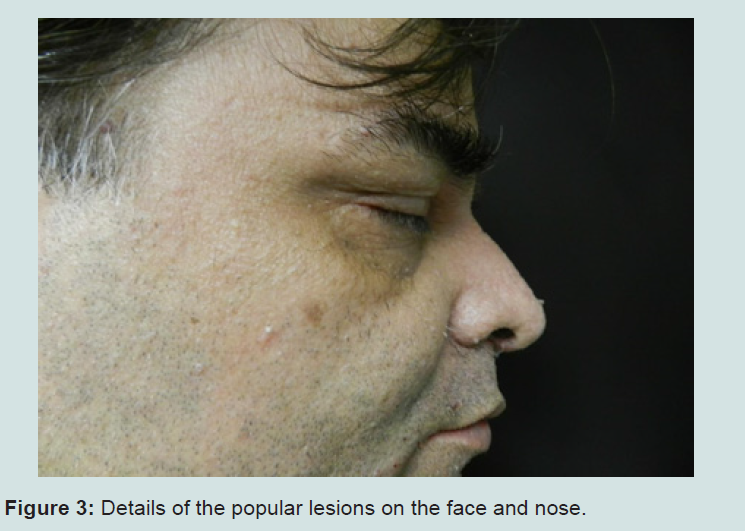
Figure 3: Details of the popular lesions on the face and nose.
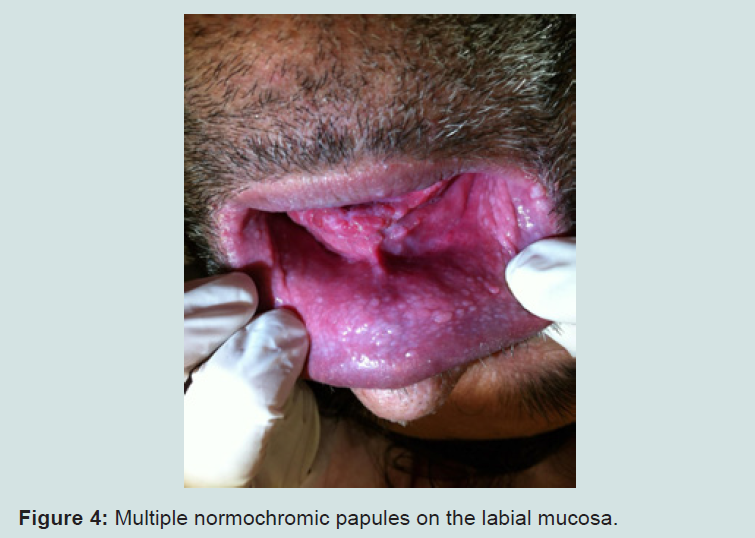
Figure 4: Multiple normochromic papules on the labial mucosa.
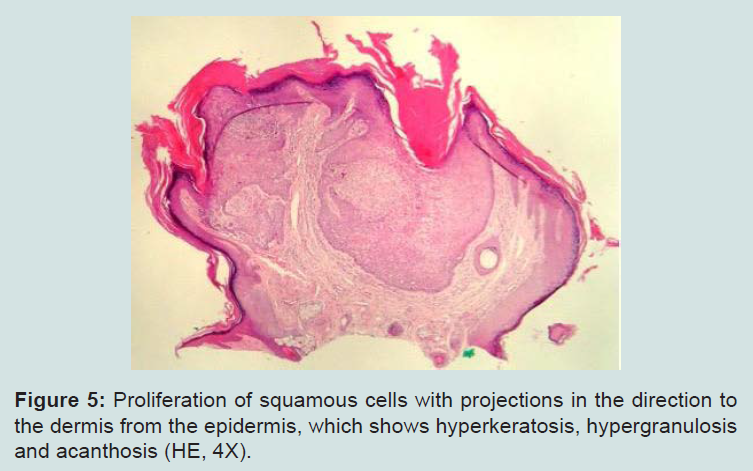
Figure 5: Functional domains and mutations in coding region of the human FOXP3 gene. FOXP3 transcript consists of 12 exons and the open reading frame is from exons 2 to 12. There are 4 potential functional domains (hollow boxes). Black starts indicate germline mutations identified from IPEX patients. Black arrows indicate somatic mutations from breast cancer patients, and solid triangles indicate somatic mutations from prostate cancer patients.
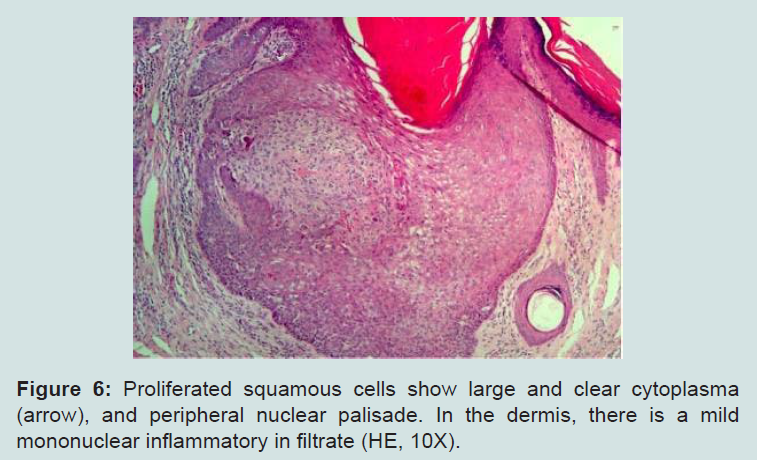
Figure 6: Functional domains and mutations in coding region of the human FOXP3 gene. FOXP3 transcript consists of 12 exons and the open reading frame is from exons 2 to 12. There are 4 potential functional domains (hollow boxes). Black starts indicate germline mutations identified from IPEX patients. Black arrows indicate somatic mutations from breast cancer patients, and solid triangles indicate somatic mutations from prostate cancer patients.
Discussion
Cowden syndrome is an autosomal dominant disorder with variable expression, caused by mutations in the germ line of the tumor suppressor gene PTEN (phosphatase and tensin homolog) of chromosomal location 10q22-23 [2]. It was described in 1963 by Lloyd & Dennis in a 20 year-old patient named Rachel Cowden [1]. Its prevalence is estimated as 1/200,000 to 1/250,000. There is predominance in Caucasians, and there seems to be preponderance in females, with the age of diagnosis varying between 13 and 65 years [3].Clinically, mutations in the PTEN gene are associated with a broad spectrum of diseases that include Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, and adult Lhermitte-Duclos disease, among others [4]. The diagnostic criteria are based on the International Cowden Consortium for diagnosis of the Cowden Syndrome (2000), which is described in Table 1 [3].
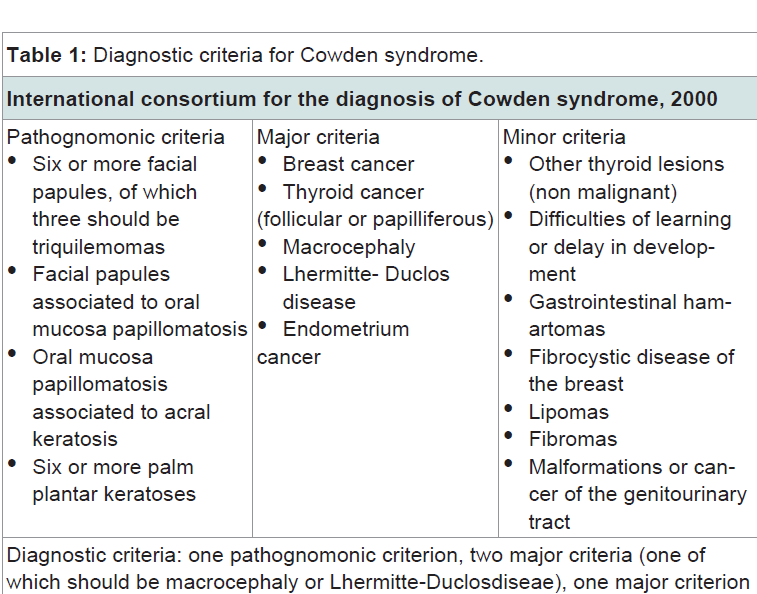
Table 1: Demographic data by sex characteristics for study population TD and ASD group.
Regarding predisposition to malignancies, breast cancer is the most common neoplasia, occurring in about 25-50% of women with the syndrome. The predominant histological subtype is ductal adenocarcinoma [7]. Thyroid cancer is the second most common cancer occurring in 3-10% of cases in both genders. The predominant subtypes are follicular or papilliferous. Additionally, benign lesions as multinodular goiter, adenomatous nodules and follicular adenomas are present in up to 75% of patients with Cowden syndrome [8]. Involvement of the gastrointestinal tract occurs in 70-85% of patients, with the presence of hamartomatous polyps predominantly in the rectum and sigmoid [9]. Lhermitte-Duclos disease, which is characterized by hamartomas of the cerebellum ganglia cells, exists separately in children, however when appearing in an adult, it is considered as pathognomonic of the syndrome’s [2,5] as observed in the diagnostic criteria in Table 1.
The key to addressing the risk of cancer is an early diagnosis, especially in cases with a positive familial history. In affected women, the clinical breast exam should be carried out every six months starting at age 25, together with mammography and annual magnetic resonance imaging [10]. Clinical thyroid examination and thyroid function tests should be performed starting at age 18, with ultrasound and aspiration puncture if there are suspected lesions. Examinations of the gastrointestinal tract and the central nervous system should be based on the presence of symptoms, since most of the lesions are benign [11]. Regarding the skin lesions that are present in most cases, treatment should be directed to their removal. In addition to surgical excision, other methods are described, as electrosurgery, cryosurgery, dermabrasion, and CO2 laser ablation [12].
Our patient presented two pathognomonic criteria (facial papules and papillomatosis of the oral mucosa), in addition to two major criteria (macrocephaly and papilliferous thyroid carcinoma) and two minor (multinodular goiter and retarded development). The patient informed there were no other cases of the syndrome in his family but it was not possible to examine any relatives or to establish a pedigree; however, the late diagnosis at age 45 caught our attention.
The diagnosis of Cowden syndrome was only made after the thyroid cancer, and despite having macrocephaly, a delayed development and exuberant mucocutaneous manifestations, which should have called attention to the diagnosis of Cowden syndrome many years before.
References
- Mukamal LV, Ferreira AF, Jacques Cde M, Amorim CA, Pineiro-Maceira J, et al. (2012) Cowden syndrome: review and report of a case of late diagnosis. Int J Dermatol 51: 1494-1499.
- Café MEM, Azulay RD (2013) Genodermatoses hiperplásicas, aplásicas, displásicas e atróficas. In: Azulay RD, Azulay DR, Azulay-Abulafia L. Azulay (eds). Dermatologia (6th edn). Rio de Janeiro: Guanabara Koogan. 857-862.
- Farooq A, Walker LJ, Bowling J, Audisio RA (2010) Cowden syndrome. Cancer Treat Rev 36: 577-583.
- Pilarski R (2009) Cowden syndrome: a critical review of the clinical literature. J Genet Couns 18: 13-27.
- Eng C (2000) Will the real Cowden syndrome please stand up: revised diagnostic criteria. J Med Genet 37: 828-830.
- Brownstein MH, Mehregan AH, Bilowski JB (1977) Trichilemmomas in Cowden’s disease. JAMA 238: 26.
- Schrager CA, Schneider D, Gruener AC, Tsou HC, Peacocke M (1998) Clinical and pathological features of breast disease in Cowden’s syndrome: an underrecognized syndrome with an increased risk of breast cancer. Hum Pathol 29: 47-53.
- Harach HR, Soubeyran I, Brown A, Bonneau D, Longy M (1999) Thyroid pathologic findings in patients with Cowden disease. Ann Diagn Pathol 3: 331-340.
- Carlson GJ, Nivatvongs S, Snover DC (1984) Colorectal polyps in Cowden’s disease (multiple hamartoma syndrome). Am J Surg Pathol 8: 763-770.
- Gustafson S, Zbuk KM, Scacheri C, Eng C (2007) Cowden syndrome. Sem Oncol 34: 428-434.
- The NCCN 1.2009 (2009) Cowden syndrome clinical practice guidelines in oncology. National Comprehensive Cancer Network.
- Wheeland RG, McGillis ST (1989) Cowden’s disease--treatment of cutaneous lesions using carbon dioxide laser vaporization: a comparison of conventional and superpulsed techniques. J Dermatol Surg Oncol 15: 1055-1059.